Whole genome sequencing and bioinformatics analysis of two Egyptian genomes
논문명: 두 이집트 게놈의 전체 게놈 시퀀싱 및 생물 정보학 분석(Whole genome sequencing and bioinformatics analysis of two Egyptian genomes)
게재저널명
Elsevier
게재년월
2018년 8월 20일
저자
MahmoudElHefnawia1SungwonJeonbc1YoungjuneBhakbcAsmaaElFikyadAhmedHoraizaJeHoonJunefHyunhoKimfJongBhakbcef
Contents
Contents
[[[hide]]]
요약
우리는 ~ 30x 시퀀싱 깊이에서 시퀀싱 된 두 개의 이집트 남성 게놈 (EGP1 및 EGP2)을 보고한다. EGP1은 470 만개의 변이체를 가지고 있으며, 198,877 개는 새로운 변이체 인 반면, EGP2는 470 만개의 변이체 중 209,109 개의 새로운 변이체를 가졌다. 두 개체의 미토콘드리아 일배체 그룹은 각각 H7b1 및 L2a1c 인 것으로 확인되었다. 또한 EGP1 (R1b)과 EGP2 (J1a2a1a2 > P58 > FGC11)의 Y haploggroup도 확인하였다. EGP1은 Leber의 유전성 시신경 병 증을 유발하는 미토콘드리아 게놈 ND4 (m.11778 G> α)의 NADH 유전자에 돌연변이가 있었다. 두 게놈에 의해 공유되는 일부 SNP는 아마도 이집트인 비만과 관련된 콜레스테롤 및 중성 지방의 증가와 관련이 있었다. 이 유전체와 아프리카 및 서아시아의 게놈을 비교하면 이집트의 조상과 유전 사에 대한 통찰력을 얻을 수 있다. 이 자료는 아프리카와 서아시아에 걸친 인간의 이동과 진화 뿐만 아니라 변이의 게놈 다양성과 기능적 분류를 더 이해하는 데 사용될 수 있다.
약어
EGP,Egyptian person;HQ,High quality;LD,Linkage disequilibrium; LHON,Leber's hereditary optic neuropathymt;DNA,Mitochondrial DNA;NGS,Next generation sequencing;rCRS,revised Cambridge reference sequence;SNP,Single nucleotide polymorphism;WGS,Whole genome sequencing;Y-STR,short tandem repeat (STR) on the Y-chromosome
키워드
전체 게놈 시퀀싱, 이집트 변종, 인류이동, 생물정보학
본문
소개
인간 게놈은 인간 진화, 다양성, 건강, 생리학 및 의학에 대한 방대한 양의 데이터를 보유하고 있다 (Lander et al., 2001). 전체 게놈 시퀀싱 (WGS) 데이터는 공통 및 희귀 장애 관련 연구와 같은 다양한 목적으로 위해 가능한 가장 심층적인 유전자 분석에 사용될 수 있다. 유전자와 그들의 다양한 변이 정보는 또한 일반적인 질병의 위험 요소를 추정하는데 효과적으로 사용될 수있다 (Bick & Dimmock, 2011; Thompson et al., 2012). 현재, 대량 병렬 차세대 시퀀싱 (NGS) 방법은 전체 인간 게놈을 분석하는 데 가장 널리 사용되는 방법이다. 게놈의 짧은 판독 값을 매핑하고 후속 변이를 호출하는 프로그램이 빠르게 개선되고 업그레이드되고 있다 (Lupski et al., 2010). 또한, 게놈 분석 비용이 매우 낮아졌으며 WGS는 영향을 받는 사람들의 게놈을 면밀히 조사함으로써 흔하지 않은 질병을 유발하는 변이체를 탐지하는데 더 보편화되고 있다(Lupski et al., 2010; Sobreira et al., 2010; Roach et al. , 2010). 예를 들어, BRCA1 및 BRCA2 유전자 변이를 가진 여성을 선별하여 유방암과 난소암의 위험을 평가하는 데 유용할 수 있다(Campau et al., 2008).
이집트 인구는 아프리카와 아시아 사이의 위치하기 때문에 다양하다. 나일 강을 따라 2 개의 긴 둑이 있으며, 아프리카에서 가장 긴 강이며 역사를 통틀어 다양한 인구를 보유하고 있다. 미라와 같은 고대 이집트 전통은 게놈을 보존하고 DNA 변이체를 분석하는 데 중요한 역할을 한다 (Paabo, 1985). 이집트 DNA는 오랫동안 연구되어왔다 (Hawass et al., 2010). 현대 북아프리카의 사람들의 DNA에 대한 연구에 따르면 그들의 유전자 빈도는 남부 유럽, 근동 및 사하라 이남 아프리카의 유전자 빈도에 중심이 된다고 밝혀졌다 (Cavalli-Sforza et al., 1994). 그러나 현대 이집트 인구의 Y 염색체의 결합되지 않는 부분의 빈도 분포는 아프리카 북부 중부 인구와 매우 유사하여 유라시아 유전 성분의 더 많은 부분을 암시한다 (Cavalli-Sforza et al., 1994; Bosch et al., 1997; Manni et al., 2002; Arredi et al., 2004; Luis et al., 2004).
Pagani 외 연구진 들은 225 명의 이집트인과 에티오피아 인을 분석 한 결과, 아프리카에서 나온 정확한 뿌리는 시나이에서 유라시아까지의 이르는 북부 지역임을 보여 주었다 (Pagani et al., 2015). Khairat, Ball 외 연구진은 미라 화 된 이집트인 중 한 명이 서아시아에서 유래한 것으로 알려진 모계혈통 인 L2 mtDNA haplogroup (Li & Durbin, 2009)과 관련이 있음을 발견했다.
여기, 우리는 높은 시퀀싱 깊이에서 두 개의 이집트 전체 게놈의 분석을 보고한다. 기능적, 유해한 돌연변이의 분석을 포함한 체계적인 게놈 분석이 이루어졌고, 우리는 아프리카와 중동지역의 인구와 비교하여 게놈 구조 및 계통수를 제공하였다.
재료 및 방법
DNA 추출 및 윤리적 승인
혈액 샘플은 부모가 각각 Delta(이집트 북부)와 Saied(이집트 남부)에서 온 두 명의 건강한 사람들로부터 추출했다. 이 연구는 IRB-REC-2011-10-003과 함께 Genome Research Foundation의 기관 검토위원회 (Institutional Review Board)의 승인을 받았으며 참가자들에 의해 서면 동의서 서명을 받았다 .
샘플 준비 및 전장 게놈 시퀀싱
제조사의 프로토콜에 따라, 유전자 JETBlood 게놈 DNA 정제 키트 (Thermo Scientific, USA)를 사용하여 혈액으로부터 게놈 DNA를 추출하였다. 삽입물 크기 400–500 insert bp의 라이브러리가 생성되었다. 게놈 DNA를 Covaris S 시리즈 (Covaris, MS, USA)를 사용하여 잘랐다. 잘라진 DNA를 A-꼬리로 말단-수리하고, 제조사의 프로토콜 (미국 캘리포니아 주 샌디에고에 소재한 Illumina의 Truseq DNA 샘플 준비 키트 v2)에 따라 페어-엔드 어댑터에 ligation시켰다. 어댑터가 부착된 단편을 2 % 아가로스 겔상에서 크기 선택하고, 520-620 bp 밴드를 제거하였다. 겔 추출 및 컬럼 정제는 제조사의 프로토콜에 따라 Minelute Gel Extraction Kit (Qiagen)를 적용하여 수행되었다. 어댑터 서열을 함유하는 ligation된 DNA 부분은 어댑터 특이적 프라이머를 사용하여 PCR을 통해 향상되었다. 라이브러리 품질 및 농도는 애질런트 2100 BioAnalyzer를 사용하여 해결되었다. 라이브러리는 Illumina의 라이브러리 정량 프로토콜에 의해 지시된 바와 같이 KAPA 라이브러리 정량 키트 (KapaBiosystems, MA, USA)를 사용하여 평가되었다. qPCR 정량에 따르면, 라이브러리를 2nM로 표준화 한 후 0.1nN NaOH를 사용하여 변성시켰다. 변성된 주형의 클러스터 증폭은 제조사의 프로토콜 (Illumina)에 따라 플로우 셀에서 완료되었다. 플로우 셀을 Illumina HiSeq2000 머신에서 페어드-엔드 시퀀싱(2 × 100100bp) 하였다. Raw 형광 이미지 및 해독된 서열을 처리하기 위해, 염기-해독 파이프 라인 (SCS (Sequencing Control Software), Illumina)이 적용되었다. 나머지 분석은 Illumina의 다운 스트림 분석 CASAVA 소프트웨어 제품군에서 유지 관리하는 FASTQ 파일에서 시작되었다. Raw 데이터는 NCBI SRA에서 액세스 번호 SRR5738871 및 SRR5738872로 액세스 할 수 있다.
참조 및 변이 탐지에 대한 판독 정렬
NGSQC 툴킷 v 2.3.3 (Patel & Jain, 2012)은 '-l 70 – s 20'옵션 (HQ = 70 %의 컷오프 판독 길이, 컷오프 품질 점수 options = 20)을 사용하여 낮은 품질의 판독 값을 필터링하는 데 적용되었다. 그 후, BWA-MEM 0.7.8 (Li & Durbin, 2009)을 기본 옵션으로 사용하여 필터링 된 판독 값을 hg19 인간 참조에 정렬했다. 그런 다음 Samtools 0.1.19 (Li et al., 2009a)를 사용하여 SAM 파일을 BAM 파일로 복원했다. PCR 중복 판독을 제거하기 위해 Picard v1.9.2 (http://broadinstitute.github.io/picard/)의 MarkDuplicate 서브 루틴을 사용했다. 또한 변이 해독의 정확성을 높이기 위해 GATK v2.3.9 (McKenna et al., 2010a)를 사용하여 IndelRealigner 및 BaseRecalibration을 수행했다. 이 변이는 GATK UnifiedGenotyper에 의해‘--heterozygosity 0.0010 -dcov 200 -stand_call_conf 30.0 –stand_emit_conf 30.0' 옵션과 함께 해독되었다.
변이의 주석 및 기능 분석
snpEff v4.3i를 사용하여 변이체의 유형 및 게놈 영역에 주석을 달았다 (Cingolani et al., 2012). 기능을 바꾸는 아미노산 변화를 야기하는 돌연변이를 예측하기 위해, PROVEAN이 사용되었다 (Choi & Chan, 2015). 이러한 단백질-손상 돌연변이는 OMIM (McKusick, 2007) 및 ClinVar 데이터베이스 (Landrum et al., 2016)로 추가로 주석이 달렸다.
계통수 제조와 ADMIXTURE 분석
먼저 PLINK 1.90 (Purcell et al., 2007)을 사용하여 Affymetrix human origin 단일 뉴클레오티드 다형성 (SNP) 판넬과 (HOSP) 데이터 (Lazaridis et al., 2014)를 두 이집트 게놈과 병합하여 591,356 개의 상 염색체 SNP를 생성했다. 우리는 LD (linkage disequilibrium)로 판넬을 정리했다. 최종 데이터 세트에는 289,287 개의 SNP가 포함되었다. 기본 교차 검증을 통해 ADMIXTURE 1.3.0 (Alexander et al., 2009)을 실행하고 조상 모집단 수 K 값을 2에서 10으로 사용했다. 아프리카와 중동 지역의 인구에 걸친 계통수를 제조하기 위해 ADMIXTURE에 대해 동일한 SNP 판넬을 갖는 pairwise 뉴클레오티드 거리를 계산했다. 그리고 우리는 pairwise pi 거리를 갖는 neighbor-joining 트리를 제조했다 (Nei & Li, 1979).
미토콘드리아 DNA와 Y 염색체 일배체 동정
미토콘드리아 일배체 그룹을 동정하기 전에, 우리는 사내 스크립트를 사용하여 짧은 hg19 레퍼런스의 chrM에 매핑된 짧은 시퀀싱 판독을 추출했다. 그런 다음 BWA-MEM (Li & Durbin, 2009)을 사용하여 판독 값을 rCRS mtDNA 레퍼런스에 매핑하고 samtools를 사용하여 샘플에 대한 동의 mtDNA 시퀀스를 만들어 냈다 (Li et al., 2009a). 각 샘플의 미토콘드리아 일배체 그룹은 MitoTool (Fan & Yao, 2013)에 의해 동정되었다. Y 염색체의 일배체 그룹은 Nevgen 예측자를 사용하여 동정되었다.
결과 및 토론
기증자
EGP1의 임상 병력은 양측 시력 상실을 나타내며, 시신경을 특이적으로 손상시키는 미토콘드리아 유전병인 Leber의 유전성 시신경 병증 (LHON)의 전형적인 증상과 일치하며 좌측 시력 상실이 더 심각한 것으로 나타났다.
표1. EGP1 안구 검사의 임상 데이터.
|
Right eye |
Left eye | |
|
Vision (cc) |
20/400 |
2/300 |
|
Color |
RE 45/12 |
LE 2.5/12 |
|
Refraction |
−4.5 sphere |
−4.5 sphere |
|
Pupils |
Sluggishly reactive, There is probably a left relative afferent pupillary defect | |
|
Ocular motility |
Ductions and Versions: Full 핵간ophthalmologia ( internuclearophthalmologia )는 없음 | |
게놈 시퀀싱 및 변이체 동정
게놈 DNA를 Hiseq 2000 플랫폼 (미국 캘리포니아 주 샌디에고 소재의 일루미나 (Illumina))에 의해 시퀀싱하여 각각 EGP1 및 EGP2에 대해 1,072,420,548 및 1,048,815,766 판독을 만들었다. NGSQC 툴킷을 사용하여 저품질 판독 값을 필터링했다 (Patel & Jain, 2012). 총 94.62Gb 및 92.68Gb의 판독 값을 유지하고 각각 BWA-MEM을 사용하여 인간 기준 hg19에 정렬시켰다 (Li & Durbin, 2009). 총 885,930,166 (99.96 %) 및 867,073,418 (99.95 %) 판독 값이 각각 EGP1 및 EGP2에 대한 참조에 매핑되어 29배 이상의 depth를 얻었다 (표 2).
Table 2. whole genome sequencing 및 mapping 요약
|
Sample ID |
EGP1 |
EGP2 |
|
Number of generated reads |
1,072,420,548 |
1,048,815,766 |
|
Number of remained reads after filtering |
946,144,204 |
926,701,466 |
|
Number of reads after removing the duplicates |
886,322,035 |
867,470,926 |
|
Number of mapped reads |
885,930,166 |
867,073,418 |
|
Mapping rate |
99.96% |
99.95% |
|
Average depth (×) |
30.4× |
29.7× |
Table 3. 각 표본의 변이 통계
|
Sample ID |
EGP1 |
EGP2 |
|
Number of homozygous SNPs |
1,486,562 |
1,473,578 |
|
Number of heterozygous SNPs |
2,702,312 |
2,815,594 |
|
Total number of SNPs |
4,188,874 |
4,289,172 |
|
Number of homozygous INDELs |
223,160 |
219,551 |
|
Number of heterozygous INDELs |
342,591 |
355,587 |
|
Total number of INDELs |
565,751 |
575,138 |
|
Number of known variants |
4,555,748 (95.82%) |
4,655,201 (95.70%) |
|
Number of total variants |
4,754,625 |
4,864,310 |
EGP1과 EGP2에서 각각 총 4,754,625개 변이와 4,864,310개의 변이를 확인했다. 4,555,748개(95.82%, EGP1)와 4,655,201개 변이는 dbSNP 데이터베이스에서 이미 보고되었으며 참고자료로 고려되었다. 또한 EGP1과 EGP2에서 4,188,874개의 SNP와 4,289,172개의 SNP를 확인했다. EGP1에는 1,486,562개의 homozygous SNP와 2,702,312개의 heterozygous SNP가 있었고, EGP2에는 표 3에 나타난 바와 같이 1,473,578개의 homozygous SNP와 2,815,594개의 heterozygous SNP가 있었다. snpEff(Cingolani et al., 2012)를 사용한 변이들의 효과에 주석을 달았고, EGP1과 EGP2 의 성적서에 대한 총 112,637의 효과와 114,964의 효과를 각각 확인했다(S1 표).
변이(variant)의 기능적 분류와 임상적 관련성
Egyptian genomes에서 non-synonymous SNPs (nsSNPs)의 가능한 기능적 효과를 파악하기 위해 computational prediction methods, the PROVEAN tool을 사용했다 (Choi & Chan, 2015). EGP1과 EGP2에서 단백질을 기능적으로 손상시키는 것으로 분류된 각각 2134와 2093개의 non-mitochondrial nsSNP를 식별하였다. 우리는 NSNP를 식별하고, 임상적 관련성을 찾기 위해 Clin Var를 사용하여 이러한 NSNP에 주석을 달았다. 이 분석은 두 명의 이집트 피험자에게서 발견된 몇몇의 nsSNP를 밝혀냈다. EGP1의 경우, herpes simplex encephalitis에 민감한 SNP (rs3775291) Leu 412 Phe in TLR3 gene가 검출되었다. 또한, NQO1의 Pro187Ser (rs1800566)는 벤젠에 노출된 후 혈액 독성의 위험이 증가하고 다른 유형의 암에 대한 민감성과 관련이 있었다 (Smith, 1999; Traver et al., 1997). EGP2는 methionine synthase (MTRR) gene에서 A66G 또는 Ile22Met으로 알려진 SNP (rs1801394)를 가졌다. 이 대체는 neural tube defect spina bifida의 위험 증가와 관련이 있으며 혈청 비타민 B12의 부족이 이 효과를 확장시켰다 (Wilson et al., 1999). 우리는 998 개의 function-altering nsSNP 가 두 이집트인에 의해 공유되었으며 16 개의 nsSNP가 여러 표현형(phenotype)과 연관되어 있음을 발견했다 (S3 표).
SNP 중 하나는 FCGR2A의 rs181274, His166Arg이며, 말라리아 루푸스 신장염에 대한 민감성과; Met580Thr in ATP6V0A4 (rs380715), renal tubular acidosis과 관련이 있다 (Smith et al., 2000). slow acetylation (Vatsis et al., 1991)과 관련된NAT2(rs1799930)의 Arg197Gln도 발견되었다. NAT2의 변이는 아세틸화의 용량과 연계되어 있으며, 모집단에 걸쳐 유전적 다양성을 가지고 있는 것으로 알려져 있다(Magalon et al., 2008). 이집트인의 약 80%는 slow acetylation를 가지고 있다(Ma et al., 2002). 우리는 또한 hyperhomocysteinemia와 관련 된 MTHFR (rs1801133, C> T)에서 Ala222Val을 발견했다. 아프리카인들은 일반적으로 CC 유전형(genotype)을 가지고 있었고 TT 유전형(genotype)은 유럽의 남쪽 방향으로 널리 퍼져 있었으며(Wilcken et al., 2003), Pathans는 같은 돌연변이를 가지고 있었다 (Ilyas et al., 2015). 또한 이 돌연변이는 이집트인들의 대장암 위험과 관련이 있다(El Awady et al., 2009).
이집트인은 평균 BMI 이상을 가지고 있으며, 대부분은 비만인 것으로 보고되었다(Ng et al., 2014). FABP2의 Thr55Ala(rs1799883)와 RP1의 Asn985Tyr(rs2293869)가 확인되었으며, 모두 콜레스테롤, 중성지방 및 아마도 비만 증가와 관련이 있었다(Georgopulos et al., 2000; Fujita et, 2003; Subraman & Chait, 2012). TYR의 Ser192Tyr(rs1042602, C > A)는 피부/헤어/눈 색깔 및 주근깨의 부재와 관련이 있었다(Sulem et al., 2007; Stokowski et al., 2007). A alle은 유럽 인구에서 긍정적인 선택을 받았다(Sulem et al., 2007). 또한, 두 개의 SNP가 위험 요소로 주석을 달았다: Pro124Leu in PLAU (rs2227564), 알츠하이머 병에 대한 민감성 및 leprosy5 와 연관된 Asn248Ser in TLR1 (rs4833095), (Schuring et al., 2009).
또한 표 4에서 볼 수 있는 2개의 이집트 샘플에서 미토콘드리아 펩타이드에 대한 9개의 아미노산 변화를 확인했다. EGP2와 EGP1에서 각각 1회 및 2회 기능변동 아미노산 변화가 예측되었다. 이것은 특히 EGP1에 Arg340Heith in ND4 (m.11778G > A)가 있었기 때문이며, 이는 eber's hereditary optic neuropathy (LHON) 과 연관되어 있다(Singh et al., 1989).
Table 4. damaging prediction of amino acid change in mitochondrial gene 결과
|
Sample |
PROTEIN_ID |
POS |
REF |
ALT |
Score |
Prediction (cutoff = −2.5) |
Gene symbol |
|
EGP1 |
ENSP00000354554 |
194 |
T |
A |
0.31 |
Neutral |
MT-CYB |
|
ENSP00000354632 |
112 |
T |
A |
−3.97 |
Deleterious |
MT-ATP6 | |
|
ENSP00000354961 |
340 |
R |
H |
−4.74 |
Deleterious |
MT-ND4 | |
|
EGP2 |
ENSP00000354499 |
254 |
I |
V |
−0.44 |
Neutral |
MT-CO1 |
|
ENSP00000354554 |
7 |
T |
I |
−2.35 |
Neutral |
MT-CYB | |
|
ENSP00000354554 |
194 |
T |
A |
0.31 |
Neutral |
MT-CYB | |
|
ENSP00000354632 |
59 |
T |
A |
−0.94 |
Neutral |
MT-ATP6 | |
|
ENSP00000354632 |
112 |
T |
A |
−3.97 |
Deleterious |
MT-ATP6 | |
|
ENSP00000355206 |
114 |
T |
A |
−1.39 |
Neutral |
MT-ND3 |
ADMIXTURE 및 계통 발생학적 분석
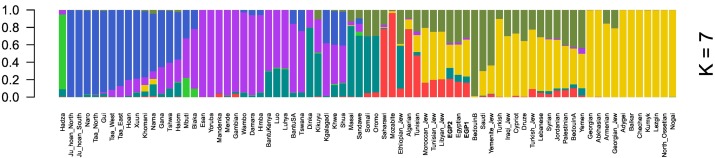
두 이집트인의 게놈 대표를 조사하기 위해, 우리는 인간 SNP 패널 (Lazaridis et al., 2014) (HOSP) (K and = samples2에서 K = 10, 그림 S1)에서 아프리카 및 중동 샘플과 함께 투여 분석을 수행했다. 우리의 이집트 게놈은 HOSP에서 이집트와 유사한 투여 구성을 가졌다. 우리는 또한 아프리카와 중동의 이집트인과 다른 인구를 비교했다. 두 이집트 게놈은 K = 7에서 중동과 아프리카 (북부와 서부)의 혼합 구성을 가졌다 (그림 1). 흥미롭게도 이집트인들은 K = 2에서 K = 4의 중서부 구성이 주요했으며 그 기원은 아프리카 외부에서 온 것으로 나타난다.
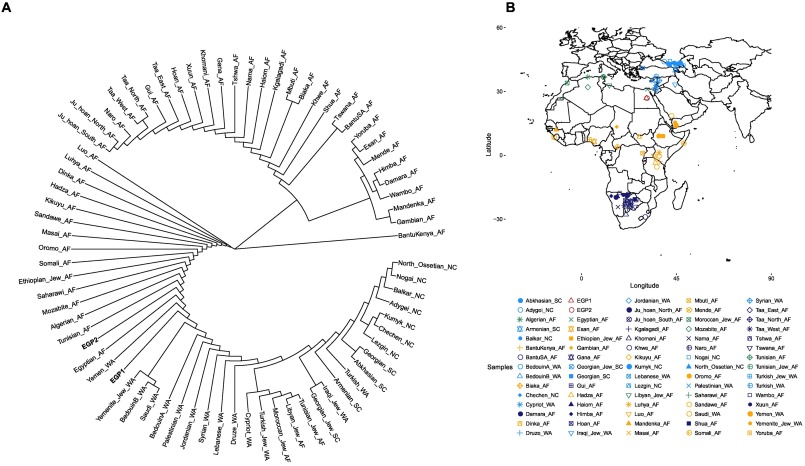
우리는 또한 HOSP의 SNPs를 기반으로 한 뉴클레오티드 별 뉴클레오티드 다양성을 계산하고 관리 분석에 사용한 아프리카와 중동의 여러 인구와 함께 두 이집트인의 계통수를 구성했다 (그림 2). EGP1과 EGP2는 중동의 자매 그룹으로 위치했다. 이것은 이집트인들이 이전에 제안한 바와 같이 훨씬 큰 유라시아 성분을 가지고 있음을 확인시켜준다 (Cavalli-Sforza et al., 1994; Bosch et al., 1997; Manni et al., 2002; Arredi et al., 2004; Luis et al., 2004) .
Download : Download high-res image (322KB)
{kind=link}
Download : Download full-size image
{kind=link}
그림 1. 아프리카와 중동의 여러 인구와 함께 이집트인의 K = 7에서 ADMIXTURE 결과.
Download : Download high-res image (910KB)
{kind=link}
Download : Download full-size image
{kind=link}
그림 2. pairwise nucleotide distance에 따른 계통 발생학적 관계.
(A) 쌍방향 뉴클레오티드 거리에 따른 계통수와 (B) 본 연구의 이집트인 2명을 포함하여 ADMIXTURE 및 계통수 구조에 사용되는 샘플의 지리적 위치.
미토콘드리아 및 Y-염색체 분석
시퀀싱 미토콘드리아 DNA (mtDNA)는 인간 인구의 모계 이동과 유전력을 이해하는 데 널리 사용된다 (Cann et al., 1987). SAMtools를 사용하여 두 이집트 게놈의 mtDNA의 합의 서열을 생성했다 (Li et al., 2009a). 우리는 MitoTool (Fan & Yao, 2013)을 사용하여 각 샘플의 미토콘드리아 haplogroup을 확인하여 EGP1에는 H7b1이 있고 EGP2에는 L2a1c가 있다는 결과를 얻었다 (표 5). H7은 대부분 근동, 코카서스, 이란, 중앙 아시아 및 Balto-Slavic 국가 (Costa et al., 2013)와 H2, H5, H7, H13 및 H20에서 발견되었다. 유럽은 현재 중동 전역에서 발견되고 있다 (van Oven & Kayser, 2009). Haplogroup L2는 아프리카 인의 약 33 %와 현재 자손에게 제공된다 (Gonder et al., 2007). L2a1c는 L2a1b와 돌연변이 16,189를 정기적으로 공유하지만, 3010 및 6663에서 그 자체의 특정 마커를 갖는다. 16,192에서의 변경은 마찬가지로 L2a1b 및 L2a1c에서 염기성이다; 동남아 및 동 아프리카에서도 나타난다. 이것은 현장에서 이 clade의 일부를 확장할 것을 권장한다. 위치 T16209C C16301T L2a1 위의 C16354T는 (Salas et al., 2002; Kivisild et al., 2004)에 의해 대부분 동 아프리카 (예 : 수단, 누비아, 에티오피아) 및 서아프리카 (예 : Turkana, Kanuri)에서 나타나는 L2a1c라는 작은 서브 클래드를 특징으로 한다.
차드 분지 (Chad Basin)에서 동아프리카 및 서아프리카 haplotypes의 몇 가지 돌연변이 진보인 4가지의 다양한 L2a1c 구성물이 구별되었다 (Kivisild et al., 2004; Cerny et al., 2007).
표 5. 2 개의 이집트 게놈의 미토콘드리아 haplotypes 및 변이체.
|
Sample ID |
mtDNA Haplotype |
Variants |
|
EGP1 |
H7b1 |
263, 309 + C, 315 + C, 750, 1438, 4769, |
|
4793, 5348, 8860, 11,778,12,351, 15,326, | ||
|
16183C, 16,189, 16,193 + C, 16519 | ||
|
EGP2 |
L2a1c |
73, 143, 146, 151, 152, 195, |
|
263, 309 + C, 315 + C, 748, 750, 769, | ||
|
1018,1438, 2416, 2706, 2789, 3010, | ||
|
3594, 4104, 4769, 6663, 7028, 7175, | ||
|
7256, 7274, 7521, 7771, 8206, 8701, | ||
|
8860, 9221, 9540, 9950, 10,115, 10,398, | ||
|
10,873, 11,719, 11,914, 11,944, 12,693, 12,705, | ||
|
13,590, 13,650, 13,803, 14,566, 14,766, 15,208, | ||
|
15,301, 15,326, 15,784, 16183C, 16,189, 16,193 + C, | ||
|
16,223, 16,278, 16,294, 16,309, 16,390, 16,519 |
Y- 염색체 반수체 그룹의 경우, FTDNA 111 Y-STR 마커에 따라 STR 마커를 식별 한 Y-STR 툴킷 (Li et al., 2009b; McKenna et al., 2010b)을 사용하여 짧은 탠덤 반복 분석을 수행했다. 마커는 Y- 할로 그룹 예측 자 (Y-DNA haplogroup Predictor – NEVGEN)에서 사용되었다. EGP1은 서유럽에서 가장 빈번하게 발생하는 것으로 알려진 R1b 일배 체형 (Myres et al., 2011)을 가지고 있으며 동유럽, 서아시아 및 일부 북아프리카 및 중앙 아시아 지역에서 온건 한 빈도로 노출되었다 ( Herrera et al., 2012). EGP2는 J1a2a1a2> P58> FGC11을 가졌다. Haplotype J1은 Caucasia, Mesopotamia, Levant 및 Arabian 반도에서 가장 많이 발견된다. J1-FGC11은 베두인과 다른 셈족 개인, 예를 들어 유대인과 아랍인 중에서 가장 널리 알려진 유전이다. 아라비아, 예멘, 에티오피아 등과 같이 불임의 토양에서 주로 관찰되기 때문에 재배와는 반대로 축산 문화와 관련 가능성이 있다 (Arredi et al., 2004; Abu-Amero et al., 2009).
결론
우리는 델타 (이집트 북부, EGP1)와 사이드 (이집트 남부, EGP2)에서 각각 2 명의 이집트인 개인의 전체 게놈을 제시한다. 우리의 분석은 이집트 게놈 이질성과 일부 변이의 기능적 특성에 대한 풍부한 데이터와 정보를 제공한다. 또한 아프리카와 중동의 여러 인구와 비교하여 두 명의 이집트인에 대한 계통 발생 정보를 제공하며, 이들의 유전 역사는 중동과 북아프리카와 동 아프리카 사이에 혼합되어 있음을 시사한다. 이것은 초기 인간 이동과 현재의 인구 다양성에 약간의 빛을 비출 수 있다. 이집트 유전학에 대한 더 큰 규모의 연구는 미래에 훨씬 더 미세한 규모와 빈도로 완전한 유전자 구성을 매핑해야한다.
다음은 이 글과 관련된 보충 데이터이다.
Download : Download Acrobat PDF file (916KB)
S1 Figure. ADMIXTURE results among populations in Africa and Middle East.
Download : Download spreadsheet (35KB)
S1 Table
Download : Download spreadsheet (7MB)
S2 Table
Download : Download spreadsheet (42KB)
S3 Table
저자 공헌
Conceptualization: ME, JB.
Data Curation: JJ, HK.
Formal analysis: SJ, YB.
Funding Acquisition: JB.
Investigation: SJ, YB, AE.
Methodology: AE, JJ.
Project Administration: JB.
Resources: ME, AE, AH.
Software: SJ, YB, AE, JJ, HK.
Supervision: JB.
Validation: SJ, YB.
Visualization: SJ.
Writing – Original Draft Preparation: ME, SJ.
Writing – Review & Editing: SJ, YB, AE, AH, JB.
감사의 말
이 작업은 범 아시아 인구 유전체 이니셔티브 (PAPGI) 컨소시엄 (http://papgi.org)의 산하에서 수행된다. 이는 범 아시아 인구의 변이를 연구하고, 이러한 변이가 이 인구와 그들의 진화에 어떻게 영향을 미치는지를 이해하는 것을 목표로 한다. 이 작업은 울산 게놈코리아프로젝트(800게놈배열)연구기금(1.180017.01)과 울산 게놈코리아프로젝트(200게놈배열)연구기금(1.180024.01)의 지원을 받았다. J. B., S. J.는 개인 게놈 정보학 연구 기금(NRF-2017M3C9A6047623)에 대한 인적 자원 개발(HRD)의 지원을 받았다.
참조
K.K. Abu-Amero, A. Hellani, A.M. Gonzalez, J.M. Larruga, V.M. Cabrera, et al.Saudi Arabian Y-chromosome diversity and its relationship with nearby regions
BMC Genet., 10 (2009), p. 59
View Record in ScopusGoogle Scholar
D.H. Alexander, J. Novembre, K. LangeFast model-based estimation of ancestry in unrelated individuals
Genome Res., 19 (2009), pp. 1655-1664
CrossRefView Record in ScopusGoogle Scholar
B. Arredi, E.S. Poloni, S. Paracchini, T. Zerjal, D.M. Fathallah, et al.A predominantly neolithic origin for Y-chromosomal DNA variation in North Africa
Am. J. Hum. Genet., 75 (2004), pp. 338-345
ArticleDownload PDFView Record in ScopusGoogle Scholar
D. Bick, D. DimmockWhole exome and whole genome sequencing
Curr. Opin. Pediatr., 23 (2011), pp. 594-600
View Record in ScopusGoogle Scholar
E. Bosch, F. Calafell, A. Perez-Lezaun, D. Comas, E. Mateu, et al.Population history of North Africa: evidence from classical genetic markers
Hum. Biol., 69 (1997), pp. 295-311
View Record in ScopusGoogle Scholar
P.M. Campeau, W.D. Foulkes, M.D. TischkowitzHereditary breast cancer: new genetic developments, new therapeutic avenues
Hum. Genet., 124 (2008), pp. 31-42
CrossRefView Record in ScopusGoogle Scholar
R.L. Cann, M. Stoneking, A.C. WilsonMitochondrial DNA and human evolution
Nature, 325 (1987), pp. 31-36
CrossRefView Record in ScopusGoogle Scholar
L.L. Cavalli-Sforza, P. Menozzi, A. PiazzaThe History and Geography of Human Genes
xi, 541, Princeton University Press, Princeton, N.J (1994)
(518 pp)
V. Cerny, A. Salas, M. Hajek, M. Zaloudkova, R. BrdickaA bidirectional corridor in the Sahel-Sudan belt and the distinctive features of the Chad Basin populations: a history revealed by the mitochondrial DNA genome
Ann. Hum. Genet., 71 (2007), pp. 433-452
CrossRefView Record in ScopusGoogle Scholar
Y. Choi, A.P. ChanPROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels
Bioinformatics, 31 (2015), pp. 2745-2747
CrossRefView Record in ScopusGoogle Scholar
P. Cingolani, A. Platts, L. Wang le, M. Coon, T. Nguyen, et al.A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3
Fly, 6 (2012), pp. 80-92
(Austin)
CrossRefView Record in ScopusGoogle Scholar
M.D. Costa, J.B. Pereira, M. Pala, V. Fernandes, A. Olivieri, et al.A substantial prehistoric European ancestry amongst Ashkenazi maternal lineages
Nat. Commun., 4 (2013), p. 2543
M.K. El Awady, A.M. Karim, L.S. Hanna, L.A. El Husseiny, M. El Sahar, et al.Methylenetetrahydrofolate reductase gene polymorphisms and the risk of colorectal carcinoma in a sample of Egyptian individuals
Cancer Biomark., 5 (2009), pp. 233-240
View Record in ScopusGoogle Scholar
L. Fan, Y.G. YaoAn update to MitoTool: using a new scoring system for faster mtDNA haplogroup determination
Mitochondrion, 13 (2013), pp. 360-363
ArticleDownload PDFView Record in ScopusGoogle Scholar
Y. Fujita, Y. Ezura, M. Emi, S. Ono, D. Takada, et al.Hypertriglyceridemia associated with amino acid variation Asn985Tyr of the RP1 gene
J. Hum. Genet., 48 (2003), pp. 305-308
CrossRefView Record in ScopusGoogle Scholar
A. Georgopoulos, O. Aras, M.Y. TsaiCodon-54 polymorphism of the fatty acid-binding protein 2 gene is associated with elevation of fasting and postprandial triglyceride in type 2 diabetes
J. Clin. Endocrinol. Metab., 85 (2000), pp. 3155-3160
View Record in ScopusGoogle Scholar
M.K. Gonder, H.M. Mortensen, F.A. Reed, A. de Sousa, S.A. TishkoffWhole-mtDNA genome sequence analysis of ancient African lineages
Mol Biol Evol, 24 (2007), pp. 757-768
CrossRefView Record in ScopusGoogle Scholar
Z. Hawass, Y.Z. Gad, S. Ismail, R. Khairat, D. Fathalla, et al.Ancestry and pathology in king Tutankhamun's family
JAMA, 303 (2010), pp. 638-647
CrossRefView Record in ScopusGoogle Scholar
K.J. Herrera, R.K. Lowery, L. Hadden, S. Calderon, C. Chiou, et al.Neolithic patrilineal signals indicate that the Armenian plateau was repopulated by agriculturalists
Eur. J. Hum. Genet., 20 (2012), pp. 313-320
CrossRefView Record in ScopusGoogle Scholar
M. Ilyas, J.S. Kim, J. Cooper, Y.A. Shin, H.M. Kim, et al.Whole genome sequencing of an ethnic Pathan (Pakhtun) from the north-west of Pakistan
BMC Genomics, 16 (2015), p. 172
T. Kivisild, M. Reidla, E. Metspalu, A. Rosa, A. Brehm, et al.Ethiopian mitochondrial DNA heritage: tracking gene flow across and around the gate of tears
Am. J. Hum. Genet., 75 (2004), pp. 752-770
ArticleDownload PDFView Record in ScopusGoogle Scholar
E.S. Lander, L.M. Linton, B. Birren, C. Nusbaum, M.C. Zody, et al.Initial sequencing and analysis of the human genome
Nature, 409 (2001), pp. 860-921
View Record in ScopusGoogle Scholar
M.J. Landrum, J.M. Lee, M. Benson, G. Brown, C. Chao, et al.ClinVar: public archive of interpretations of clinically relevant variants
Nucleic Acids Res., 44 (2016), pp. D862-868
CrossRefView Record in ScopusGoogle Scholar
I. Lazaridis, N. Patterson, A. Mittnik, G. Renaud, S. Mallick, et al.Ancient human genomes suggest three ancestral populations for present-day Europeans
Nature, 513 (2014), pp. 409-413
CrossRefView Record in ScopusGoogle Scholar
H. Li, R. DurbinFast and accurate short read alignment with burrows-wheeler transform
Bioinformatics, 25 (2009), pp. 1754-1760
CrossRefView Record in ScopusGoogle Scholar
H. Li, B. Handsaker, A. Wysoker, T. Fennell, J. Ruan, et al.The sequence alignment/map format and SAMtools
Bioinformatics, 25 (2009), pp. 2078-2079
CrossRefView Record in ScopusGoogle Scholar
H. Li, B. Handsaker, A. Wysoker, T. Fennell, J. Ruan, et al.The sequence alignment/map format and SAMtools
Bioinformatics, 25 (2009), pp. 2078-2079
CrossRefView Record in ScopusGoogle Scholar
J.R. Luis, D.J. Rowold, M. Regueiro, B. Caeiro, C. Cinnioglu, et al.The Levant versus the horn of Africa: evidence for bidirectional corridors of human migrations
Am. J. Hum. Genet., 74 (2004), pp. 532-544
ArticleDownload PDFView Record in ScopusGoogle Scholar
J.R. Lupski, J.G. Reid, C. Gonzaga-Jauregui, D. Rio Deiros, D.C. Chen, et al.Whole-genome sequencing in a patient with Charcot-Marie-tooth neuropathy
N. Engl. J. Med., 362 (2010), pp. 1181-1191
View Record in ScopusGoogle Scholar
M.K. Ma, M.H. Woo, H.L. McLeodGenetic basis of drug metabolism
Am. J. Health Syst. Pharm., 59 (2002), pp. 2061-2069
CrossRefView Record in ScopusGoogle Scholar
H. Magalon, E. Patin, F. Austerlitz, T. Hegay, A. Aldashev, et al.Population genetic diversity of the NAT2 gene supports a role of acetylation in human adaptation to farming in Central Asia
Eur. J. Hum. Genet., 16 (2008), pp. 243-251
CrossRefView Record in ScopusGoogle Scholar
F. Manni, P. Leonardi, A. Barakat, H. Rouba, E. Heyer, et al.Y-chromosome analysis in Egypt suggests a genetic regional continuity in Northeastern Africa
Hum. Biol., 74 (2002), pp. 645-658
CrossRefView Record in ScopusGoogle Scholar
A. McKenna, M. Hanna, E. Banks, A. Sivachenko, K. Cibulskis, et al.The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data
Genome Res., 20 (2010), pp. 1297-1303
CrossRefView Record in ScopusGoogle Scholar
A. McKenna, M. Hanna, E. Banks, A. Sivachenko, K. Cibulskis, et al.The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data
Genome Res., 20 (2010), pp. 1297-1303
CrossRefView Record in ScopusGoogle Scholar
V.A. McKusickMendelian inheritance in man and its online version, OMIM
Am. J. Hum. Genet., 80 (2007), pp. 588-604
ArticleDownload PDFCrossRefView Record in ScopusGoogle Scholar
N.M. Myres, S. Rootsi, A.A. Lin, M. Jarve, R.J. King, et al.A major Y-chromosome haplogroup R1b Holocene era founder effect in central and Western Europe
Eur. J. Hum. Genet., 19 (2011), pp. 95-101
CrossRefView Record in ScopusGoogle Scholar
M. Nei, W.H. LiMathematical model for studying genetic variation in terms of restriction endonucleases
Proc. Natl. Acad. Sci. U. S. A., 76 (1979), pp. 5269-5273
CrossRefView Record in ScopusGoogle Scholar
M. Ng, T. Fleming, M. Robinson, B. Thomson, N. Graetz, et al.Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the global burden of disease study 2013
Lancet, 384 (2014), pp. 766-781
ArticleDownload PDFView Record in ScopusGoogle Scholar
M. van Oven, M. KayserUpdated comprehensive phylogenetic tree of global human mitochondrial DNA variation
Hum. Mutat., 30 (2009), pp. E386-394
View Record in ScopusGoogle Scholar
S. PaaboMolecular cloning of ancient Egyptian mummy DNA
Nature, 314 (1985), pp. 644-645
CrossRefView Record in ScopusGoogle Scholar
L. Pagani, S. Schiffels, D. Gurdasani, P. Danecek, A. Scally, et al.Tracing the route of modern humans out of Africa by using 225 human genome sequences from Ethiopians and Egyptians
Am. J. Hum. Genet., 96 (2015), pp. 986-991
ArticleDownload PDFView Record in ScopusGoogle Scholar
R.K. Patel, M. JainNGS QC toolkit: a toolkit for quality control of next generation sequencing data
PLoS One, 7 (2012), Article e30619
S. Purcell, B. Neale, K. Todd-Brown, L. Thomas, M.A. Ferreira, et al.PLINK: a tool set for whole-genome association and population-based linkage analyses
Am. J. Hum. Genet., 81 (2007), pp. 559-575
ArticleDownload PDFCrossRefView Record in ScopusGoogle Scholar
J.C. Roach, G. Glusman, A.F. Smit, C.D. Huff, R. Hubley, et al.Analysis of genetic inheritance in a family quartet by whole-genome sequencing
Science, 328 (2010), pp. 636-639
CrossRefView Record in ScopusGoogle Scholar
A. Salas, M. Richards, T. De la Fe, M.V. Lareu, B. Sobrino, et al.The making of the African mtDNA landscape
Am. J. Hum. Genet., 71 (2002), pp. 1082-1111
ArticleDownload PDFView Record in ScopusGoogle Scholar
R.P. Schuring, L. Hamann, W.R. Faber, D. Pahan, J.H. Richardus, et al.Polymorphism N248S in the human toll-like receptor 1 gene is related to leprosy and leprosy reactions
J. Infect. Dis., 199 (2009), pp. 1816-1819
G. Singh, M.T. Lott, D.C. WallaceA mitochondrial DNA mutation as a cause of Leber's hereditary optic neuropathy
N. Engl. J. Med., 320 (1989), pp. 1300-1305
View Record in ScopusGoogle Scholar
M.T. SmithBenzene, NQO1, and genetic susceptibility to cancer
Proc. Natl. Acad. Sci., 96 (1999), pp. 7624-7626
View Record in ScopusGoogle Scholar
A.N. Smith, J. Skaug, K.A. Choate, A. Nayir, A. Bakkaloglu, et al.Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing
Nat. Genet., 26 (2000), pp. 71-75
View Record in ScopusGoogle Scholar
N.L. Sobreira, E.T. Cirulli, D. Avramopoulos, E. Wohler, G.L. Oswald, et al.Whole-genome sequencing of a single proband together with linkage analysis identifies a Mendelian disease gene
PLoS Genet., 6 (2010), Article e1000991
R.P. Stokowski, P.V. Pant, T. Dadd, A. Fereday, D.A. Hinds, et al.A genomewide association study of skin pigmentation in a South Asian population
Am. J. Hum. Genet., 81 (2007), pp. 1119-1132
ArticleDownload PDFCrossRefView Record in ScopusGoogle Scholar
S. Subramanian, A. ChaitHypertriglyceridemia secondary to obesity and diabetes
Biochim. Biophys. Acta, 1821 (2012), pp. 819-825
ArticleDownload PDFView Record in ScopusGoogle Scholar
P. Sulem, D.F. Gudbjartsson, S.N. Stacey, A. Helgason, T. Rafnar, et al.Genetic determinants of hair, eye and skin pigmentation in Europeans
Nat. Genet., 39 (2007), pp. 1443-1452
CrossRefView Record in ScopusGoogle Scholar
R. Thompson, C.J. Drew, R.H. ThomasNext generation sequencing in the clinical domain: clinical advantages, practical, and ethical challenges
Adv. Protein Chem. Struct. Biol., 89 (2012), pp. 27-63
ArticleDownload PDFView Record in ScopusGoogle Scholar
R. Traver, D. Siegel, H. Beall, R.M. Phillips, N. Gibson, et al.Characterization of a polymorphism in NAD (P) H: quinone oxidoreductase (DT-diaphorase)
Br. J. Cancer, 75 (1997), pp. 69-75
CrossRefView Record in ScopusGoogle Scholar
K.P. Vatsis, K.J. Martell, W.W. WeberDiverse point mutations in the human gene for polymorphic N-acetyltransferase
Proc. Natl. Acad. Sci. U. S. A., 88 (1991), pp. 6333-6337
CrossRefView Record in ScopusGoogle Scholar
B. Wilcken, F. Bamforth, Z. Li, H. Zhu, A. Ritvanen, et al.Geographical and ethnic variation of the 677C>T allele of 5,10 methylenetetrahydrofolate reductase (MTHFR): findings from over 7000 newborns from 16 areas world wide
J. Med. Genet., 40 (2003), pp. 619-625
View Record in ScopusGoogle Scholar
A. Wilson, R. Platt, Q. Wu, D. Leclerc, B. Christensen, et al.A common variant in methionine synthase reductase combined with low cobalamin (vitamin B 12) increases risk for spina bifida
Mol. Genet. Metab., 67 (1999), pp. 317-323