Genome-wide data from two early Neolithic East Asian individuals dating to 7700 years ago. Siska, V., Jones, E. R., Jeon, S., Bhak, Y., Kim, H. M., Cho, Y. S., . . . Manica, A. (2017). Sci Adv, 3(2), e1601877
Sci Adv. 2017 Feb; 3(2): e1601877.
Published online 2017 Feb 1. doi: 10.1126/sciadv.1601877
PMCID: PMC5287702
PMID: 28164156
Contents
Contents
[[[hide, V., Jones, E. R., Jeon, S., Bhak, Y., Kim, H. M., Cho, Y. S., . . . Manica, A. (2017). Sci Adv, 3(2), e1601877|hide]]]
- 1Contents
- 27700 년 전으로 거슬러 올라가는 초기 신석기 시대 동아시아 두 개인의 게놈 전체 데이터
- 3서론
- 4결과
- 5토의
- 6재료 및 방법론
- 7SUPPLEMENTARY MATERIALS
Contents[edit]
[[[hide]]]
7700 년 전으로 거슬러 올라가는 초기 신석기 시대 동아시아 두 개인의 게놈 전체 데이터[edit]
[Author&cauthor=true&cauthor_uid=28164156 Veronika Siska],1,* [Author&cauthor=true&cauthor_uid=28164156 Eppie Ruth Jones],1,2 [Author&cauthor=true&cauthor_uid=28164156 Sungwon Jeon],3 [Author&cauthor=true&cauthor_uid=28164156 Youngjune Bhak],3 [Author&cauthor=true&cauthor_uid=28164156 Hak-Min Kim],3 [Author&cauthor=true&cauthor_uid=28164156 Yun Sung Cho],3 [Author&cauthor=true&cauthor_uid=28164156 Hyunho Kim],4 [Author&cauthor=true&cauthor_uid=28164156 Kyusang Lee],5 [Author&cauthor=true&cauthor_uid=28164156 Elizaveta Veselovskaya],6 [Author&cauthor=true&cauthor_uid=28164156 Tatiana Balueva],6 [Author&cauthor=true&cauthor_uid=28164156 Marcos Gallego-Llorente],1 [Author&cauthor=true&cauthor_uid=28164156 Michael Hofreiter],7 [Author&cauthor=true&cauthor_uid=28164156 Daniel G. Bradley],2 [Author&cauthor=true&cauthor_uid=28164156 Anders Eriksson],1 [Author&cauthor=true&cauthor_uid=28164156 Ron Pinhasi],8,*† [Author&cauthor=true&cauthor_uid=28164156 Jong Bhak],3,4,*†‡ and [Author&cauthor=true&cauthor_uid=28164156 Andrea Manica]1,*†
Author information Article notes Copyright and License information Disclaimer
This article has been cited by other articles in PMC.
관련 데이터
요약[edit]
고대 게놈은 Holocene 선사 시대, 특히 서부 유라시아의 신석기 적 전이에 대한 우리의 이해에 혁명을 가져왔다. 대조적으로, 동아시아는 신석기 전환이 근동에서 시작된 후 약 3 천년 동안 독립적으로 일어난 핵심 지역을 대표함에도 불구하고 지금까지 거의 주목을 받지 못했다. 우리는 러시아와 한국의 국경에 위치한 동아시아에 위치한 초기 신석기 시대 동굴 유적지 (약 770 만 년 전) 인 Devil’s Gate의 두 수렵·채집인의 게놈 전체 데이터를 보고한다. 이 두 개체는 모두 Amur Basin의 모든 가까운 퉁구스 언어, 특히 Ulchi와 지리적으로 가까운 현대 인구와 유전적으로 가장 유사하다. 근처 현대 근대 인구와 유사하고 Ulchi의 낮은 수준의 추가 유전 물질은 Holocene 동안 이 지역에서 높은 수준의 유전자 연속성을 의미하며 이는 유럽에서 보고 된 것과 현저하게 대조되는 패턴이다.
키워드: 고대 유전학, 동아시아, 신석기 시대, 러시아 극동, 인구 유전학
서론[edit]
서아시아의 고대 게놈은 12 ~ 8 천년 전에 선 농업 사냥꾼 수집가와 초기 농민 사이에 어느 정도의 유전적 연속성을 보여주었다 (ka) (1, 2). 대조적으로, 남동 및 중부 유럽에 대한 연구에 따르면 8.5 ~ 7 ka 기간 동안 근동 출신의 신석기 시대 농부들이 Mesolithic 사냥꾼 채집자를 대체했다. 그런 다음 중후반 신석기 시대와 신석기 시대의 일부 지역에서 지역 수렵·채집인의 혈통의 점진적인 "재발"과 청동기 시대의 출현과 일치하여 나중에 ~ 5.5 ka의 아시아 스텝의 주요 기여가 이어진다(3–5). 수백 개의 부분 고대 게놈이 이미 서열 분석된 서부 유라시아와 비교할 때, 동아시아는 아메리카대륙의 식민지화라는 맥락에서 주목받아온 시베리아 북극 벨트를 제외하고는 고대 DNA 연구에 의해 등한시되어 왔다. 아메리카 (6, 7). 그러나 동아시아는 농업에 대한 의존도가 유라시아 서부와 다른 방향으로 바뀌었기 때문에 매우 흥미로운 지역이다. 후자 지역에서는 도기, 농업 및 축산이 밀접한 관련이 있었다. 반면 러시아 극동, 일본, 한국의 초기 신석기 시대 사회는 도기류와 바구니류를 10.5 ~ 15 ka에 제조 및 사용하기 시작했지만 가축 작물과 가축은 수천 년 후에 도착했다 (8, 9). 현재 동아시아 게놈이 부족하기 때문에, 우리는 점진적인 신석기의 전이와 서유라시아에서 발생한 것과는 개별적으로 발생한 유럽에서 발견된 실제 이주나 인구 연속성과 관련된 문화적 확산의 정도를 알지 못한다.
결과[edit]
샘플, 시퀀싱 및 진위[edit]
동아시아 신석기에 대한 지식의 이러한 차이를 메우기 위해 5 개의 초기 신석기 매장 (DevilsGate1, 0.059-fold coverage; DevilsGate2, 0.023-fold coverage) 및 DevilsGate3, DevilsGate4 및 DevilsGate5, < 러시아 극동 프리 모리에 지역의 악마의 문 (Chertovy Vorota) 동굴의 단일 직업 단계에서 0.001 배 적용) (중국 및 북한과의 국경에 근접) (보충 자료 참조) 이 지역 9.4 ~ 7.2 ka로 거슬러 올라가며, 사람의 유골은 ~ 7.7 ka로 추정되며, 고대 섬유의 세계 최초 증거 중 일부를 포함한다 (10) Devil’s Gate에 사는 사람들은 농업의 증거가 없는 hunter-fisher-gatherers 였다; 야생 식물의 섬유는 직물 생산의 주요 원료였다 (10). 시퀀싱 범위가 가장 높은 DevilsGate1 및 DevilsGate2가 가장 높은 두 샘플에 대한 분석에 중점을 둔다. 더 높은 적용 범위 (DevilsGate1)를 가진 개체의 미토콘드리아 게놈은 haplogroup D4에 할당될 수 있다; 이 haplogroup은 동아시아의 현재 인구에서 발견되며 (11) 일본 북부의 Jomon 골격에서도 발견된다 (2). 다른 개인 (DevilsGate2)의 경우 M 지점 (D4가 속한)의 구성원 만 설정할 수 있다. DevilsGate1 및 DevilsGate2 각각에 대한 haplogroup-defining 위치의 비합의 기반에 대해 미토콘드리아 DNA (mtDNA) 서열에서 불일치 호출 횟수로 추정된 오염은 {0.87 % [95 % 신뢰 구간 (CI), 0.28 ~ 2.37 %] 및 0.59 % (95 % CI, 0.03 ~ 3.753 %)}로 낮았다.
더 높은 범위의 게놈에서 schmutzi (12)를 사용하여 DevilsGate1은 또한 낮은 오염 수준을 제공한다 [1 % (95 % CI, 0 ~ 2 %); 보충 자료 참조].
오염의 잠재적 혼란 영향에 대한 추가 점검으로, 우리는 가장 중요한 분석 [외부 f3 점수 및 주요 성분 분석 (PCA)]이 사후 손상 증거 (최소 3의 PMD 점수)를 나타내는 판독만 사용하여 정성적으로 복제되었는지 확인했다. ) (13), 이 후자의 결과는 낮은 적용 범위 (DevilsGate1의 경우 0.005X 및 DevilsGate2의 경우 0.001X)로 인해 높은 수준의 노이즈를 가졌다.
현대인과의 관계[edit]
우리는 악마의 문에서 현대 유라시아의 큰 패널과 고대 게놈을 출판 한 사람들을 비교했다 (그림 1A) (4, 5, 14–17). PCA (18)와 감독되지 않은 군집 접근법 인 ADMIXTURE (19)에 근거하여, 두 개인은 모두 악마의 문이 위치한 지역인 아무르 분지의 인구에서 볼 수 있는 현대적 변동 범위에 속한다 (그림 1). 이 지역은 오늘날 단일 언어 가족 (텅스텐)의 사용자들이 거주하고 있다. 이 결과는 서구 유라시아에서 관찰된 것과 대조적이다. 서구 유라시아에서는 여러 주요 개입 이동 파 때문에 비슷한 나이의 수렵·채집인들이 현대 유전자 변이를 벗어난다 (3, 20). 우리는 그들이 아프리카 외집단으로부터 벗어났기 때문에 현대나 고대 인구의 a Devil’s Gate 개인과 현대의 X 사이에 공유된 유전적 표류의 양을 측정하는 f3 (아프리카; 데빌 게이트, X) 형식의 외부 집단 f3 통계를 사용하여 악마의 문과 현대 아무르 분지 집단 사이의 친화성을 확인했다. Devil 's Gate와 동일한 지역에 거주하는 현대 인구는 우리의 고대 게놈에 대해 가장 높은 유전 친화력을 가지고 있으며 (그림 2), 지리적 거리가 증가함에 따라 친화도가 점차 감소한다 (r2 = 0.756, F1,96 = 301, P < 0.001; 그림 3), 중립 드리프트와 일치하여 간단한 거리별 격리 패턴으로 이어진다. 전통적으로 악마의 문과 지리적으로 매우 가까운 곳에 살고 러시아에서 샘플링 된 아무르 분지에서 온 툰구스어를 사용하는 유일한 인구인 울치 (우리 패널의 다른 모든 튜구스어 사용자는 중국 출신)는 유 전적으로 패널에서 가장 비슷한 인구다. Devil 's Gate에 높은 친화력을 보이는 다른 인구는 Oroqen과 Hezhen이며, 울치와 마찬가지로 아무르 분지의 툰 구스 스피커와 현대 한국인과 일본인이다. 악마의 문과 지리적으로 거리가 멀다는 점을 고려할 때 (그림 3) 아메리카 원주민 인구는 이전에 시베리아와 다른 북아시아 인구와의 관계에 따라 이 사이트의 샘플과 유별나게 유사하다 (7).
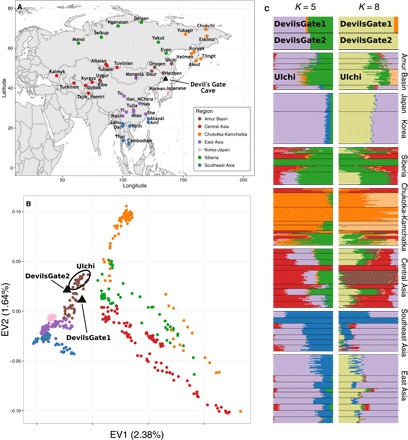
지역 참조 패널, PCA 및 ADMIXTURE 분석.
(A) Devil’s Gate (검은 삼각형)의 위치 및 분석의 지역 패널을 형성하는 현대 인구의 위치를 보여주는 아시아 지도. (B) (A)에 표시된 동아시아 및 중앙 아시아의 현대 인구 지역 패널에 의해 정의 된 한 두 가지 주요 구성 요소의 플롯과 Devil’s Gate (검은 색 삼각형)의 두 샘플이 그 위에 투영된다 (18). (C) ADMIXTURE 분석 (19) : K = 5 (가장 낮은 교차 검증 오류) 와 K = 8 (악마의 게이트 별 군집)에 대해 Devil 's Gate 및 지역 패널 에서 수행했다.

Outgroup f3 통계
Outgroup f3은 아프리카 아웃그룹(Khomani)과 관련하여 Devil’s Gate(검은색 삼각형은 샘플링 위치를 표시함)와 현대 모집단 사이의 공유 표류를 측정한다. (A) 전 세계의지도. (B) 그림과 같이 지역별로 색으로 구분 된 Devil’s Gate와 함께 가장 많이 표류 된 15개의 모집단이다. 1.오차 막대는 1 SE를 나타낸다.
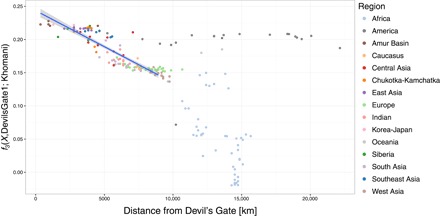
outgroup f3 통계의 공간 패턴.
outgroup f3(X, Devil 's Gate; Khomani)와 Devils Gate1 및 모든 단일 뉴클레오티드 다형성 (SNP)을 사용하여 Devil 's Gate에서 떨어진 거리와의 관계. 상관 관계를 계산할 때 Devil’s Gate에서 최대 9000km 떨어진 인구가 고려되었다. 고려 된 최고 거리는 500km 간격으로 가장 높은 Pearson 상관 관계를 얻기 위해 선택되었다. 최적의 선형 적합 (r2 = 0.772, F1,108 = 368.4, P <0.001)은 파란색 선으로 표시되며 95 % CI는 음영 영역으로 표시된다.
아시아의 고대 게놈과의 관계[edit]
이전에 발표 된 고대 게놈에는 Devil’s Gate에 대한 유전적 친화성이 표시되어 있지 않았다. outgroup f3 통계량의 상위 50 개 인구는 모두 현대적이며, 다른 고대 게놈은 지리적으로나 시간적으로 Devil’s Gate과 매우 거리가 먼 것으로 예상된다. 이 고대 유전체 중에서 Devil’s Gate에 가장 가까운 것은 청동기 이후의 스테페족과 유럽의 중석기 수렵채집가 이다. 그러나 이 유전체는 같은 지역의 현대 인구의 유전체보다 Devil’s Gate 유전체에 더 가깝지 않다. (예 : Tuvinian, Kalmyk, Russian 또는 Finnish). 지리적으로 Devil’s Gate에 가장 가까운 두 개의 고대 게놈 인 Ust'-Ishim (~ 45 ka)와 Mal'ta (MA1, 24 ka)도 유전 적 친화력이 높지 않다. 아마도 둘 다 훨씬 더 이른 시기부터 연대가 되어 있기 때문 일 것이다. 이 중 MA1은 유전적으로 Devil’s Gate에 더 가깝지만 다른 모든 동아시아 사람들과 마찬가지로 Devil’s Gate에서 먼 거리에 있다 (그림 S14에서 S16). Ust'-Ishim에서도 비슷한 패턴이 발견되는데, 이는 Devil’s Gate를 포함하여 모든 아시아인들과 거리가 똑같다. 이것은 족보의 기본 위치와 일치한다 (그림 S17 ~ S19).
Devil’s Gate와 Ulchi의 연속성[edit]
Devil 's Gate는 많은 분석에서 아무르 강의 현대 인간의 유전 변이 범위에 속하고 Ulchi에 높은 유전 친화도를 나타내기 때문에 이 지역의 유전 연속성의 정도를 조사했다. Ulchi에서 추가 유전 물질의 신호를 찾기 위해 혼합 f3 통계를 사용하여 Devil 's Gate와 다른 현대 인구를 혼합물로 모델링했다. 가능한 현대적인 소스로 구성된 대규모 패널에도 불구하고 Ulchi는 더 이상의 기여없이 Devil 's Gate만으로 가장 잘 대표된다 (혼합물 f3은 유의미한 부정적인 결과를 초래하지 않았다; 표 S3 및 S4). 혼합물 f3은 병목 현상과 같은 인구 통계학적 사건의 영향을 받을 수 있기 때문에 Devil 's Gate는 D 통계량(African outgroup, X; Ulchi, Devil 's Gate)을 사용하여 Ulchi와 함께 clade를 형성했는지 테스트했다. 전 세계적으로 많은 현대인 집단이 유의하게 0이 아닌 이는 ADMI FORDS 분석에서 Ulchi에 대한 추가 성분과 함께 연속성이 절대적이지 않음을 시사한다. 그러나 DNA 분해와 낮은 적용 범위로 인한 Devil’s Gate 시퀀스에서 오류율이 높을수록 추정 된 연속성 수준도 감소 할 수 있다는 점에 유의해야 한다. Ulchi와 Devil 's 주민 사이의 연속성 수준을 현대 유럽인과 European hunter-gatherers간의 지속성 수준을 비교하기 위해 우리는 그들의 조상 비율을 ADMIXTURE에서 유추 한 대로 비교했다. Ulchi의 Devil 's Gate 관련 조상의 비율은 모든 European hunter-gatherers 관련 조상보다 현저히 높은 것으로 나타났다 (최고의 평균 hunter-gatherers 관련 구성 요소를 가진 5 명의 유럽 인구에 대해 100 부트 스트랩 복제에서 P <0.01).
이 결과는이 영역에서 비교적 높은 수준의 연속성을 나타낸다. Ulchi는 아마도 Devil 's Gate (또는 유 전적으로 매우 가까운 인구)의 후손 일 가능성이 있지만, 이 지역의 인구들 사이의 지리적, 유전 적 연결성은이 현대 인구들 또한 관련 현대 인구와의 연관성이 높다는 것을 의미한다. 유럽과 비교했을 때, 이러한 결과는 신석기 시대 초기부터 인구가 크게 증가하지 않았던 지난 ~ 770 만 년 동안 동북 아시아에서 더 높은 수준의 유전자 연속성을 보여준다.
한국인과 일본인의 남부 및 북부 유전 물질[edit]
Devil’s Gate와 더 먼 남쪽에 사는 현대 일본인과 한국인 사이의 밀접한 유전적 친화력도 관심사이다. 고고학 (21)과 유전자 분석 (22-25)을 바탕으로, 현대 일본인은 Jomon 문화의 hunter-gatherers (16-3ka)와 중국 남부의 양쯔강 하구에서 습식 쌀 농사를 가져온 Yayoi 문화 (3 ~ 1.7ka) 이민자들의 혼합 사건에서 유래하면서 이중적 기원을 가지고 있다는 주장이 제기 돼 왔다.
북부 홋카이도 섬의 Jomon 지역에서 구할 수 있는 몇 안 되는 고대 mtDNA 샘플은 현대 일본 인구, 특히 아이누족(Anuu)과 류큐아족(Ryukyuans) , 남부 시베리안(예: degey and Ulchi)족에 존재하는 특정한 하플로타입(N9b와 M7a, G1도 검출됨)의 enrichment을 보여준다. Davil's Gate(D4 및 M)에서 추출한 샘플의 mtDNA 하플로그 그룹은 Jomon 샘플에도 포함되어 있지만 가장 일반적인 샘플은 아니다(N9b 및 M7a). 최근에 두 Jomon 표본에서 나온 핵유전자 데이터는 이중 기원 가설을 확인시켰으며, 오늘날 동아시아인이 다양화되기 전에 Jomon 이 분열했음을 암시했다.(28) 우리는 혼합 f3 통계를 사용하여 현대 아시아 인과 Devil’s Gate을 포함하여 가능한 모든 쌍의 출처를 혼합한 것으로 현대 일본어로 모델링하여 북부 및 남부 유전자 구성 요소를 복구 할 수 있는지 조사했다. 가장 명확한 신호는 대만, 중국 남부 및 베트남의 Devil’s Gate와 현대 인구의 조합에 의해 주어졌으며 (그림 4), 각각 hunter-gatherer와 농업가 구성 요소를 나타낼 수 있었다. 그러나 이러한 점수는 겨우 유의수준에 불과했고 (-3 <z <-2), 일부 현대 pair도 significance 임계 값에 도달하지 않더라도 음의 점수를 주었다 (z 점수는 -1.9만큼 낮음; 참고). 보충 자료). 한국인의 기원은 그다지 주목을 받지 못했다. 또한, 본토의 위치 때문에 한국인들은 역사적으로 이웃 주민들과 더 많은 접촉을 경험했을 것이다. 그러나 이들의 게놈은 게놈 전체의 SNP 데이터에서 일본의 것과 유사한 특성을 보이며 (29), 북아시아 및 남아시아 mtDNA (30)와 Y 염색체 반수체 그룹 (30, 31) 모두를 보유하는 것으로 나타났다. 불행히도, Devil 's Gate에서 나온 낮은 적용 범위와 작은 샘플 크기는 성분이 2 차 접촉 (혼합) 또는 연속 분화에서 비롯된 것인지, 그리고 어떤 혼합 사건을 일으켰는지 조사하기 위해 혼합물 계수의 신뢰성이 있는 추정이나 연계 불균형 기반 방법의 사용을 방지했다.

혼합물 f3 통계.
혼합물 f3는 현대 한국인과 일본인을 X와 Y의 두 인구의 혼합으로 나타내지며, 그림 1과 같이 지역별로 색상이 구분되어 있다. (A) 한국인에 대한 f3 점수가 가장 낮은 30 쌍. 유의미한 (z <-2) 값. (B) 네 쌍 모두 일본인에 대해 유의미한 (z <-2) 음수 점수를 제공한다. 오차 막대는 1 SE를 나타낸다.
관심 있는 표현형[edit]
우리의 표본의 낮은 적용 범위는 표현형의 관심 특성과 관련된 대부분의 SNP를 직접 관찰할 수는 없지만 현대 인구에 근거한 대치가 일부 정보를 제공할 수 있다. 우리는 이전에 고대 유럽 표본 (5, 16, 17)에 대한 유전자형 확률 (GP)을 추정하는 데 사용된 것과 같은 대치 법을 사용하여 가장 높은 적용 범위 DevilsGate1을 가진 게놈에 집중했다. DevilsGate1은 갈색 눈 (HERC2의 rs12913832; GP, 0.905)을 가지고 있었으며, 결정될 수 있는 경우 동아시아에서 공통적인 색소 관련 변형이 있었다 (섹션 S11 참조) (32). 그녀는 Ectodysplasin A 수용체 (rs3827760; GP, 0.865)를 암호화하여 EDAR 유전자에 유래된 돌연변이의 적어도 하나의 사본을 가지고 있는 것으로 보이며, 이는 곧고 굵은 모발 (33)의 확률을 높이고 삽 모양의 앞니를 증가시킨다 (34). 그녀는 유당 내성에 대한 가장 흔한 유라시아 돌연변이가 거의 없었으며 (rs4988235, LCT 유전자; GP> 0.999) (35) 알코올 홍조 (rs671, ALDH2 유전자; GP, 0.847)로 고통받지 않았을 것이다 (36). 따라서, 적어도 유전적 기초가 알려져 있는 표현형 특성과 관련하여, 이 7.7ky 동안이 영역에서 어느 정도의 표현형 연속성이 존재하는 것으로 보인다.
토의[edit]
러시아 극동의 데빌 스 게이트 (Devil 's Gate)의 초기 신석기 시대 동아시아 두 명의 게놈 전체 데이터를 분석함으로써, 우리는 적어도 지난 7700 년 동안이 지역에서 높은 수준의 유전자 연속성을 보여줄 수 있었다. 현대 인구가 여전히 많은 사냥꾼-어부 관행에 의존하는 이 지역의 추운 기후 조건은 동남 및 중부 유럽의 사례에 기록된 것처럼 외생 농업 인구에 의한 명백한 연속성과 주요 유전자 전환의 부족에 대한 설명을 제공할 것이다. 따라서 현지 사냥꾼 수집가들이 원래의 라이프 스타일에 식량 생산 관행을 점진적으로 추가한 것은 그럴듯해 보인다. 그러나 흥미로운 점은 유럽에서 매우 높은 위도에서 최근까지도 비슷한 생존 방식이 여전히 중요한 신석기 시대의 확장은 대륙의 남부와 비교하여 현대 인구가 비록 약화되었지만 그럼에도 중요한 유전적 특징을 남겼다는 것이다. 따라서 우리의 고대 게놈은 서부 유라시아와 비교하여 동아시아의 신석기 전이 동안 질적으로 다른 인구 역사에 대한 증거를 제공하여 이전 지역에서 더 강한 유전 적 연속성을 시사한다. 이 결과는 동아시아 신석기 시대에 대한 추가 연구를 장려하는데, 이는 초기 동식물의 유전 데이터 (이상적으로는 동남아시아의 습식 벼 경작지 근처의 지역)와 집중 농업 전의 인구 구조를 정량화하기 위해 얻은 다른 지역 샘플인 더 넓은 범위의 사냥꾼-수집가 표본으로부터 크게 수혜를 받는다고 볼 수 있다.
재료 및 방법론[edit]
실험 설계[edit]
샘플 준비 및 시퀀싱[edit]
실험적 설계 샘플 준비 및 시퀀싱 분자 분석은 아일랜드 더블린의 Trinity College의 고대 DNA 시설에서 수행되었다. 샘플을 제조하고, Gamba 등의 방법에 따라 실리카 컬럼 기반 프로토콜을 사용하여 DNA를 추출하였으며 (17), 이는 Yang et al. (37)의 연구에 기반하였다. 제1 및 제2 용해 완충제 (17)로부터 추출된 DNA를 라이브러리 제조에 사용하였고, 이는 Gamba et al. (17)에 의해 설명된 대로 Meyer 및 Kircher (38)의 프로토콜의 수정된 방식에 기초하였다. 라이브러리는 Illumina MiSeq 또는 HiSeq 플랫폼에서 시퀀싱 되었다 (자세한 내용 및 시퀀싱 통계는 보충 자료 및 방법 참조).
데이터 처리 및 정렬[edit]
단일 말단 시퀀싱 데이터의 경우 cutadapt (39)를 사용하여 읽기 끝에서 어댑터 시퀀스를 잘라내어 어댑터와 읽기 간에 1 개의 기본 쌍 (bp)만 겹치게 했다. 짝지은 말단 데이터의 경우 leeHom (40)을 사용하여 어댑터를 트리밍했다. leeHom은 --ancientdna 옵션을 사용하여 실행되었으며, 겹쳐진 짝지은 말단 읽기는 병합되었다. 겹칠 수 없는 짝지은 말단 읽기의 경우 읽기 1의 데이터 만 다운 스트림 분석에 사용되었다. 시드 영역이 비활성화된 상태에서 Burrows-Wheeler 정렬 도구 (BWA) (41)를 사용하여 미토콘드리아 서열이 수정된 케임브리지 참조 서열 (National Center of Biotechnology Information accession 번호 NC_012920.1)로 대체된 휴먼 게놈의 GRCh37 빌드에 대해 판독이 정렬되었다. 서로 다른 시퀀싱 실험의 읽기는 Picard MergeSamFiles (http://picard.sourceforge.net/)를 사용하여 병합되었고 SAMtools (42)를 사용하여 클론 읽기를 제거했다. 최소 판독 길이는 30 bp이며, 높은 커버리지 (0.01 이상) 샘플인 DevilsGate1 및 DevilsGate2의 경우 GATK (Genome Analysis Toolkit)의 RealignerTargetCreator 및 IndelRealigner를 사용하여 indel을 재정렬했다 (43). SAMtools (42)를 사용하여 매핑 품질이 30 미만인 판독 값을 필터링하고 mapDamage 2.0 (44)을 사용하여 판독 값을 다시 조정하여 손상된 염기의 품질을 낮추어 분석 (44)에 관한 고대 DNA 손상 관련 오류의 영향을 줄였다. 평균 게놈 피복 깊이는 bedtools의 genomecov 기능을 사용하여 계산되었다 (45).
결과의 진위성 및 오염 추정[edit]
DevilsGate3, DevilsGate4 및 DevilsGate5에 대한 모든 판독 값을 사용하여 분자 손상 패턴 및 판독 길이 분포를 평가하였다. DevilsGate1 및 DevilsGate2의 읽기 중 일부는 50bp 단일 엔드 시퀀싱에서 파생되었으므로 잘린 읽기 사용을 피하기 위해 다음 분석에서 150bp 짝지은 말단 시퀀싱으로 시퀀싱 된 읽기만 고려했다 (라이브러리 ID MOS5A.E1 DevilsGate1 및 DevilsGate2의 경우 MOS4A.E1). MapDamage 2.0 (44)은 고대 DNA의 전형적인 분자 손상 패턴을 평가하는 데 사용되었다. 우리는 낮은 범위의 데이터에 손상을 주는 것처럼 보이는 진정한 돌연변이가 생기는 편견을 피하기 위해 MapDamage를 적용하지 않고 데이터를 사용하여 모든 결과를 복제했다. 자세한 내용은 보충 자료 및 방법을 참조. 가장 높은 범위의 시료인 DevilsGate1 및 DevilsGate2에 대한 미토콘드리아 오염 비율을 평가했다. 이는 품질이 20 이상인 염기를 사용하여 일 배 그룹 정의 위치 (DevilsGate1의 경우 D12 및 DevilsGate2의 경우 M)에서 비동의성 베이스의 백분율을 평가하여 계산되었다. 또한 DevilsGate1의 미토콘드리아 오염을 추정하기 위해 베이지안 최대 사후 알고리즘 (12)을 사용하는 도구인 schmutzi를 사용했다. 마지막으로, 우리는 사후 손상의 증거 (최소 3의 PMD 점수)를 보여주는 판독만 사용하여 가장 강력한 결과 (외부 f3 점수 및 PCA)를 산출 한 분석을 복제했다 (13). 자세한 내용은 보충 자료 및 방법을 참조.
통계 분석[edit]
미토콘드리아 일 배체 그룹 결정 및 분자 성별 결정[edit]
차세대 시퀀싱 데이터 (ANGSD)의 분석을 사용하여 Devils Gate 1 및 Devils Gate 2에 대해 미토콘드리아 동의 시퀀스를 생성했다 (46). 해독 위치는 커버리지 깊이가 3 이상이어야 하고 품질이 20 이상인 베이스만 고려되었다. 생성된 FASTA 파일을 haplogroup 결정을 위해 HAPLOFIND (47)에 업로드하고 GATK DepthOfCoverage (43)를 사용하여 적용 범위를 계산했다. 할당된 haplogroup을 정의하는 돌연변이도 수동으로 확인했다. Skoglund et al.의 연구에 기술된 스크립트를 사용하여 분자 성별을 할당하였다. (48). 자세한 내용은 보충 자료 및 방법을 참조.
SNP 추출 및 참조 패널과의 병합[edit]
우리의 샘플을 현대적이고 고대적인 인간의 유전자 변이와 비교하기 위해, 우리는 SAMtools 1.2 (42)를 사용하여 Human Origins (HO) reference panel (591,356 positions) (49) 과 겹치는 위치에서 hg19 reference FASTA file 를 사용하여 SNPs를 추출했다. 베이스(Bases)는 최소 매핑 품질 30과 기본 품질 20이 요구되었고, 모든 triallelic SNP는 폐기되었다. 우리의 낮은 커버리지(low coverage)는 infer diploid genotypes을 추론하기에 충분한 정보를 제공하지 않기 때문에, 커버리지의 깊이와 비례하는 확률로 베이스가 선택되었다. 이 대립 유전자(allele)는 homozygous diploid genotype을 형성하기 위해 복제되었으며, 이 유전형은 해당 SNP 위치(48)에서 개인을 나타내는 데 사용되었다. 이 SNP 추출 방식(지금부터 비례 방식이라고 함)은 고대 개인으로 이어지는 혈통에 drift의 appearance을 인위적으로 증가시키겠지만, 이러한 드리프트는 특정한 방향으로 예상되지 않으므로, 인구 관계(3)에 대한 추론을 편향해서는 안 된다. DevilsGate1의 총 35,903개의 position과 DevilsGate2의 14,739개의 position들은 적어도 한 개의 high-quality read로 커버되었다.
그런 다음 DevilsGate1과 DevilsGate2에 대한 SNP 데이터는 HO 패널의 최신 게놈을 포함하는 참조 패널과 병합되었고, 고대 게놈[이 데이터 세트는 Jones외(5)에 의해 설명됨]과 Personal Genome Project Korea (http://opengenome.net/)에서 PLINK 1.07(50)를 사용하여 45개의 한국 게놈을 추가로 선택되었다. 샘플 ID, 모집단 및 논문 전체에서 사용된 그룹화를 포함하여 확장 데이터 표 S1에서 추가 샘플 정보를 사용할 수 있다. 마지막으로 위의 모든 데이터에 대한 transversion-only version이 모든 T를 C로, G를 A로 변환하여 생성되었다. 이 대체 데이터 세트(alternative data set)는 고대 DNA 손상에서 비롯되는 잠재적 편견이 우리의 결론에 영향을 미치지 않음을 확인하기 위해 사용되었다.
이후 분석 (outgroup and admixture f3 statistics, PCA, and ADMIXTURE analysis),에서는 감소 된 정보 함량으로 인한 변환 전용 데이터( transversion-only data )의 증가 된 노이즈를 제외하고, 모든 돌연변이 또는 변환(transversions)만을 사용한 결과는 질적으로 유사 하였다. 따라서 본문에서는 모든 돌연변이와 비례법이라고 하는 기본 추출법을 사용하여 결과만을 보고한다(어떤 주어진 위치를 포함하는 리드(read)에서 무작위로 리드(read)를 균일하게 선택). 우리는 outgroup f3 및 D 통계에 대한 아프리카 outgroup으로서 Khomani San을 사용하여 결과를 제시하지만, 다른 인구(Yoruba, the Mbuti, the Dinka)는 동등한 결과를 나타냈다. 자세한 내용은 보충 자료 및 방법을 참조.
인구유전분석[edit]
PCA는 두 개의 서로 다른 참조 패널(그림 1 및 확장 데이터 표 S1)로 수행되었으며, 이는 존스 외 연구진(5)의 연구로부터 동시대와 고대 개인으로 구성된 전 세계 패널의 하위 세트였다. 분석은 lsqproject와 함께 EIGENSOFT 6.0.1 smartpca (18) 및 정규화 옵션 설정(normalization options on), the outlier removal option off 및 r2> 0.2가 제거 된 linkage disequilibrium의 각 쌍에서 하나의 SNP를 사용하여 수행되었다.고대의 표본들은 현대 인구에 의해 정의된 주요 성분들에 투영되었다. 자세한 내용과 결과는 보충 자료 및 방법을 참조.
ADMI COLATES 버전 1.23(19)을 사용하여 클러스터링분석(clustering analysis0을 수행하였다.linkage disequilibrium SNP는 parameters –indep-pairwise 200 25 0.5인 PLINK 1.07를 사용하여 얇아졌으며, 그 결과 분석을 위한 334,359개의 SNP세트가 발생했다(91,379 transversions). 글로벌 패널의 경우 K = 2 ~ 20 개의 클러스터와 지역 패널의 K = 2 ~ 10 개의 클러스터는 10 개의 독립적 인 실행을 사용하여 탐색되었으며, 각 K에서 서로 다른 무작위 시드로 5배 교차 검증했다. 글로벌 패널의 경우 K = 18, 지역 패널(동아시아와 중앙아시아)의 경우 K = 5에서 최소 교차 검증 오류가 발견되었지만, 오류는 이미 글로벌 패널의 경우 K = 9 부근에서 정점에 도달하기 시작하여 거의 개선되지 않는다는 것을 시사한다. 게다가, 지역 패널의 결과는 동아시아 인구를 위한 글로벌 패널의 결과와 대체로 유사했다. 추론된 조상 성분의 모집단 수준 빈도(population-level frequencies)를 비교하기 위해, Devil’s Gate에서 나온 모든 SNP와 MapDamage-treated samples에서 ADMIXTURE 실행의 유추 조상 구성 요소를 사용하여 100개의 부트스트랩 추정치와 함께 각 인구에 대한 SNP와 개인에 대한 부트스트래핑을 동시에 수행했다. 앨런토프트 외 연구진(16)과 시코라 외 연구진(51)의 논리에 따라 해당 우도 함수를 수치적으로 최대화하여 각각의 부트스트랩 복제품을 조상 성분에 투영했다. 자세한 내용과 결과는 보충 자료 및 방법 및 확장 데이터 그림을 참조. S1에서 S8까지.
D 통계(52)와 f3 통계(49, 53)를 사용하여 ADMIXTULS 패키지(49)의 qpDstat(D 통계) 및 qp3PopTest(f3 통계) 프로그램을 사용하여 샘플 간의 관계를 공식적으로 평가하였다.이 프로그램들에 의해 게놈의5-centimorgan chunks가 넘는 블록 잭나이프(block jackknife)를 사용하여 유의성을 평가하였고, z 점수가 2보다 크거나 혼합물 f3 점수가 -2보다 작을 경우 통계가 유의하다고 간주되었다. 이들은 각각 0.046과 0.023의 P 값에 근사적으로 대응한다. 자세한 내용과 결과는 보충 자료 및 방법을 참조.
관심 표현형(Phenotypes of interest)[edit]
우리는 유라시아 인구에서 선택되고 있는 것으로 알려진 몇몇 loci 을 포함하여 highest-coverage 샘플인 DevilsGate1에 대한 관심 표현 유형을 조사했다. 우리 샘플의 품질이 낮기 때문에, 우리는 1000 Genomes Project (26개의 다른 인구)의 단계별 게놈(phased genomes)을 포함하는 참조 패널을 이용하여 BEAGLE (54)를 이용하여 유전자형을 귀속시켰다. 감바 외 연구진(17)의 연구에 따라, GATK UnifiedGenotypeper(43)는1000 Genomes Project의 3단계에서 SNP 사이트의 유전자형 가능성을 추출하는 데 사용되었다. 스패닝 시퀀스 데이터(spanning sequence data)가 없는 위치 및 관측된 유전자형이 탈아미노화(17)에 의해 설명될 수 있는 위치에 대해 등우도가 설정되었다. 직접 유전자형이 없는 마커의 유전자형을 추정하기 위해 10회 반복을 사용하여 관심 지역으로부터 최소 1 Mb의 상·하류를 귀속시켰다. 자세한 내용과 결과는 보충 자료 및 방법 및 확장 데이터 표 S9를 참조.
Supplementary Material[edit]
http://advances.sciencemag.org/cgi/content/full/3/2/e1601877/DC1:
감사의 말 [edit]
자금지원: V.S.는 Gates Cambridge Trust의 지원을 받았다. R.P.는 European Research Council (ERC)의 지원을 받아 ADNABIOARC(263441)를 시작으로 2014년 1월부터 2016년 12월까지 Irish Research Council Advanced Research Project Grant가 지원했다. M.H.는 ERC Consolidator Grant 310763 “GeneFlow.”의 지원을 받았다. 이 작업은 울산과학기술원 연구기금(1.140113.01)으로 J.B에 지원했다. 이 작업은 또한 J.B. 및 Y.S.C.에 대한 민사 기술 협력 프로그램의 연구 기금 (14-BR-SS-03)에 의해 지원되었다. M.G.L.는 생명공학 및 생물학 연구 위원회 박사 연수 파트너십 학생쉽의 지원을 받았다. A.M.과 A.E.는 ERC Consolidator Grant 647787 “LocalAdaptation”의 지원을 받았다. D.G.B.는ERC Investigator grant 295729-CodeX의 지원을 받았다.
저자 공헌: E.V., T.B. 및 R.P.가 샘플을 입수하여 고고학적 맥락을 제공하였다; E.R.J., H.-M.K., Y.S.C., H.K., K.L.가 실험을 수행하였다; V.S, E.R.J, S.J, Y.B, Y.C, M.G.-L, J.B. 및 A.M.에서 유전자 데이터를 분석하였다.;
V.S.M.H.D.G.B.A.E.R.P.J.B.A.M.은 모든 공저자의 입력으로 원고를 작성했다.
경쟁 관심사: 저자들은 경쟁적 이해관계가 없다고 선언한다.
데이터 및 자료 가용성: 논문의 결론을 평가하는 데 필요한 모든 데이터는 논문 및/또는 보충 자료에 수록되어 있다. 이 논문과 관련된 추가 자료는 저자들에게 요청될 수 있다. FASTA 형식의 원시 리드와 Devil’s Gate의 모든 5개 고대 샘플에 대한 bamfiles로 정렬된 리드는 European Nucleotide Archive accession code PRJEB14817에서 사용할 수 있다.
SUPPLEMENTARY MATERIALS[edit]
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/2/e1601877/DC1
Supplementary Materials and Methods
fig. S1. Calibrated age range of the two human specimens from Devil’s Gate (OxCal version 4.2.4).
fig. S2. Damage patterns for samples from Devil’s Gate.
fig. S3. Sequence length distribution for samples from Devil’s Gate.
fig. S4. Outgroup f3 statistics on PMDtools-filtered data.
fig. S5. PCA on all SNPs using the worldwide panel.
fig. S6. PCA on transversion SNPs using the worldwide panel.
fig. S7. PCA on all SNPs using the regional panel.
fig. S8. PCA on transversion SNPs using the regional panel.
fig. S9. ADMIXTURE analysis cross-validation (CV) error as a function of the number of clusters (K) for the regional panel using all SNPs (top row) or transversions only (bottom row) and with (left column) or without (right column) MapDamage treatment.
fig. S10. ADMIXTURE analysis CV error as a function of the number of clusters (K) for the world panel using all SNPs (top row) or transversions only (bottom row) and with (left column) or without (right column) MapDamage treatment.
fig. S11. Outgroup f3 scores of the form f3(X, MA1; Khomani), with modern populations and selected ancient samples (DevilsGate1, DevilsGate2, Ust’-Ishim, Kotias, Loschbour, and Stuttgart), using all SNPs, with f3 > 0.15 displayed.
fig. S12. D scores of the form D(X, Khomani; MA1, DevilsGate1), with all modern populations in our panel and selected ancient samples, using all SNPs.
fig. S13. D scores of the form D(X, Khomani; MA1, DevilsGate1), with all modern populations in our panel and selected ancient samples, using all SNPs.
fig. S14. Outgroup f3 scores of the form f3(X, Ust’-Ishim; Khomani), with modern populations and selected ancient samples (MA1, Kotias, Loschbour, and Stuttgart), using all SNPs, with f3 > 0.15 displayed.
fig. S15. D scores of the form D(X, Khomani; Ust’-Ishim, DevilsGate1), with all modern populations in our panel and selected ancient samples, using all SNPs.
fig. S16. D scores of the form D(X, Khomani; Ust’-Ishim, DevilsGate2), with all modern populations in our panel and selected ancient samples, using all SNPs.
fig. S17. Comparison of Devil’s Gate–related ancestry in the Ulchi and European hunter-gatherer–related ancestry in European populations.
fig. S18. Comparison of Devil’s Gate–related ancestry in the Ulchi and Early European farmer–related ancestry in European populations.
fig. S19. Comparison of Devil’s Gate–related ancestry in the Ulchi and Bronze Age Steppe–related ancestry in European populations.
table S1. Details of sample preparation and sequencing.
table S2. mtDNA contamination estimates.
table S3. Admixture f3(Source1, Source2; Target) for the Ulchi with z < −1 using all SNPs.
table S4. Admixture f3(Source1, Source2; Target) for the Ulchi with z < −1 using only transversion SNPs.
table S5. Admixture f3(Source1, Source2; Target) for the Sardinians using all SNPs and showing the 10 most significantly negative pairs.
table S6. Admixture f3(Source1, Source2; Target) for the Lithuanians using all SNPs and showing the 10 most significantly negative pairs.
extended data fig. S1. Results from ADMIXTURE analysis using the regional panel, all SNPs, and MapDamage treatment on samples from Devil’s Gate and setting the number of clusters to K = 2 to 10.
extended data fig. S2. Results from ADMIXTURE analysis using the regional panel, transversion SNPs, and MapDamage treatment on samples from Devil’s Gate and setting the number of clusters to K = 2 to 10.
extended data fig. S3. Results from ADMIXTURE analysis using the regional panel, all SNPs, and no MapDamage treatment on samples from Devil’s Gate and setting the number of clusters to K = 2 to 10.
extended data fig. S4. Results from ADMIXTURE analysis using the regional panel, transversion SNPs, and no MapDamage treatment on samples from Devil’s Gate and setting the number of clusters to K = 2 to 10.
extended data fig. S5. Results from ADMIXTURE analysis using the total panel, all SNPs, and MapDamage treatment on samples from Devil’s Gate and setting the number of clusters to K = 2 to 20.
extended data fig. S6. Results from ADMIXTURE analysis using the total panel, transversion SNPs, and MapDamage treatment on samples from Devil’s Gate and setting the number of clusters to K = 2 to 20.
extended data fig. S7. Results from ADMIXTURE analysis using the total panel, all SNPs, and no MapDamage treatment on samples from Devil’s Gate and setting the number of clusters to K = 2 to 20.
extended data fig. S8. Results from ADMIXTURE analysis using the total panel, transversion SNPs, and no MapDamage treatment on samples from Devil’s Gate and setting the number of clusters to K = 2 to 20.
extended data table S1. Sample information.
extended data table S2. ADMIXTURE proportions.
extended data table S3. Outgroup f3 statistics for Devil’s Gate.
extended data table S4. Outgroup f3 and space.
extended data table S5. Outgroup f3 for MA1 and Ust’-Ishim.
extended data table S6. D scores for MA1 and Ust’-Ishim.
extended data table S7. D scores for the Ulchi.
extended data table S8. Admixture f3 for the Koreans and the Japanese.
extended data table S9. Phenotypes of interest.
REFERENCES AND NOTES[edit]
1. Lazaridis I., Nadel D., Rollefson G., Merrett D. C., Rohland N., Mallick S., Fernandes D., Novak M., Gamarra B., Sirak K., Connell S., Stewardson K., Harney E., Fu Q., Gonzalez-Fortes G., Jones E. R., Alpaslan Roodenberg S., Lengyel G., Bocquentin F., Gasparian B., Monge J. M., Gregg M., Eshed V., Mizrahi A.-S., Meiklejohn C., Gerritsen F., Bejenaru L., Blüher M., Campbell A., Cavalleri G., Comas D., Froguel P., Gilbert E., Kerr S. M., Kovacs P., Krause J., McGettigan D., Merrigan M., Andrew Merriwether D., O’Reilly S., Richards M. B., Semino O., Shamoon-Pour M., Stefanescu G., Stumvoll M., Tönjes A., Torroni A., Wilson J. F., Yengo L., Hovhannisyan N. A., Patterson N., Pinhasi R., Reich D., Genomic insights into the origin of farming in the ancient Near East. Nature 536, 419–424 (2016). [PMC free article] [PubMed] [Google Scholar]
2. Gallego-Llorente M., Connell S., Jones E. R., Merrett D. C., Jeon Y., Eriksson A., Siska V., Gamba C., Meiklejohn C., Beyer R., Jeon S., Cho Y. S., Hofreiter M., Bhak J., Manica A., Pinhasi R., The genetics of an early Neolithic pastoralist from the Zagros, Iran. Sci. Rep. 6, 31326 (2016). [PMC free article] [PubMed] [Google Scholar]
3. Lazaridis I., Patterson N., Mittnik A., Renaud G., Mallick S., Kirsanow K., Sudmant P. H., Schraiber J. G., Castellano S., Lipson M., Berger B., Economou C., Bollongino R., Fu Q., Bos K. I., Nordenfelt S., Li H., de Filippo C., Prüfer K., Sawyer S., Posth C., Haak W., Hallgren F., Fornander E., Rohland N., Delsate D., Francken M., Guinet J.-M., Wahl J., Ayodo G., Babiker H. A., Bailliet G., Balanovska E., Balanovsky O., Barrantes R., Bedoya G., Ben-Ami H., Bene J., Berrada F., Bravi C. M., Brisighelli F., Busby G. B. J., Cali F., Churnosov M., Cole D. E. C., Corach D., Damba L., van Driem G., Dryomov S., Dugoujon J.-M., Fedorova S. A., Gallego Romero I., Gubina M., Hammer M., Henn B. M., Hervig T., Hodoglugil U., Jha A. R., Karachanak-Yankova S., Khusainova R., Khusnutdinova E., Kittles R., Kivisild T., Klitz W., Kučinskas V., Kushniarevich A., Laredj L., Litvinov S., Loukidis T., Mahley R. W., Melegh B., Metspalu E., Molina J., Mountain J., Näkkäläjärvi K., Nesheva D., Nyambo T., Osipova L., Parik J., Platonov F., Posukh O., Romano V., Rothhammer F., Rudan I., Ruizbakiev R., Sahakyan H., Sajantila A., Salas A., Starikovskaya E. B., Tarekegn A., Toncheva D., Turdikulova S., Uktveryte I., Utevska O., Vasquez R., Villena M., Voevoda M., Winkler C. A., Yepiskoposyan L., Zalloua P., Zemunik T., Cooper A., Capelli C., Thomas M. G., Ruiz-Linares A., Tishkoff S. A., Singh L., Thangaraj K., Villems R., Comas D., Sukernik R., Metspalu M., Meyer M., Eichler E. E., Burger J., Slatkin M., Pääbo S., Kelso J., Reich D., Krause J., Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 513, 409–413 (2014). [PMC free article] [PubMed] [Google Scholar]
4. Haak W., Lazaridis I., Patterson N., Rohland N., Mallick S., Llamas B., Brandt G., Nordenfelt S., Harney E., Stewardson K., Fu Q., Mittnik A., Bánffy E., Economou C., Francken M., Friederich S., Garrido Pena R., Hallgren F., Khartanovich V., Khokhlov A., Kunst M., Kuznetsov P., Meller H., Mochalov O., Moiseyev V., Nicklisch N., Pichler S. L., Risch R., Rojo Guerra M. A., Roth C., Szécsényi-Nagy A., Wahl J., Meyer M., Krause J., Brown D., Anthony D., Cooper A., Werner Alt K., Reich D., Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 522, 207–211 (2015). [PMC free article] [PubMed] [Google Scholar]
5. Jones E. R., Gonzalez-Fortes G., Connell S., Siska V., Eriksson A., Martiniano R., McLaughlin R. L., Gallego Llorente M., Cassidy L. M., Gamba C., Meshveliani T., Bar-Yosef O., Müller W., Belfer-Cohen A., Matskevich Z., Jakeli N., Higham T. F. G., Currat M., Lordkipanidze D., Hofreiter M., Manica A., Pinhasi R., Bradley D. G., Upper Palaeolithic genomes reveal deep roots of modern Eurasians. Nat. Commun. 6, 8912 (2015). [PMC free article] [PubMed] [Google Scholar]
6. Raghavan M., DeGiorgio M., Albrechtsen A., Moltke I., Skoglund P., Korneliussen T. S., Grønnow B., Appelt M., Christian Gulløv H., Max Friesen T., Fitzhugh W., Malmström H., Rasmussen S., Olsen J., Melchior L., Fuller B. T., Fahrni S. M., Stafford T. Jr, Grimes V., Renouf M. A. P., Cybulski J., Lynnerup N., Lahr M. M., Britton K., Knecht R., Arneborg J., Metspalu M., Cornejo O. E., Malaspinas A.-S., Wang Y., Rasmussen M., Raghavan V., Hansen T. V. O., Khusnutdinova E., Pierre T., Dneprovsky K., Andreasen C., Lange H., Hayes M. G., Coltrain J., Spitsyn V. A., Götherström A., Orlando L., Kivisild T., Villems R., Crawford M. H., Nielsen F. C., Dissing J., Heinemeier J., Meldgaard M., Bustamante C., O’Rourke D. H., Jakobsson M., Gilbert M. T. P., Nielsen R., Willerslev E., The genetic prehistory of the New World Arctic. Science 345, 1255832 (2014). [PubMed] [Google Scholar]
7. Raghavan M., Steinrücken M., Harris K., Schiffels S., Rasmussen S., DeGiorgio M., Albrechtsen A., Valdiosera C., Ávila-Arcos M. C., Malaspinas A.-S., Eriksson A., Moltke I., Metspalu M., Homburger J. R., Wall J., Cornejo O. E., Víctor Moreno-Mayar J., Korneliussen T. S., Pierre T., Rasmussen M., Campos P. F., de Barros Damgaard P., Allentoft M. E., Lindo J., Metspalu E., Rodríguez-Varela R., Mansilla J., Henrickson C., Seguin-Orlando A., Malmström H., Stafford T. Jr, Shringarpure S. S., Moreno-Estrada A., Karmin M., Tambets K., Bergström A., Xue Y., Warmuth V., Friend A. D., Singarayer J., Valdes P., Balloux F., Leboreiro I., Vera J. L., Rangel-Villalobos H., Pettener D., Luiselli D., Davis L. G., Heyer E., Zollikofer C. P. E., Ponce de León M. S., Smith C. I., Grimes V., Pike K.-A., Deal M., Fuller B. T., Arriaza B., Standen V., Luz M. F., Ricaut F., Guidon N., Osipova L., Voevoda M. I., Posukh O. L., Balanovsky O., Lavryashina M., Bogunov Y., Khusnutdinova E., Gubina M., Balanovska E., Fedorova S., Litvinov S., Malyarchuk B., Derenko M., Mosher M. J., Archer D., Cybulski J., Petzelt B., Mitchell J., Worl R., Norman P. J., Parham P., Kemp B. M., Kivisild T., Tyler-Smith C., Sandhu M. S., Crawford M., Villems R., Smith D. G., Waters M. R., Goebel T., Johnson J. R., Malhi R. S., Jakobsson M., Meltzer D. J., Manica A., Durbin R., Bustamante C. D., Song Y. S., Nielsen R., Willerslev E., Genomic evidence for the Pleistocene and recent population history of native Americans. Science 349, aab3884 (2015). [PMC free article] [PubMed] [Google Scholar]
8. Kuzmin Y., The Paleolithic-to-Neolithic transition and the origin of pottery production in the Russian Far East: A geoarchaeological approach. Archaeol. Ethnol. Anthropol. Eurasia 4, 16–26 (2003). [Google Scholar]
9. Z. Chi, H. Hsiao-chun, The Encyclopedia of Global Human Migration (Blackwell Publishing Ltd., 2013). [Google Scholar]
10. Kuzmin Y. V., Keally C. T., Jull A. J. T., Burr G. S., Klyuev N. A., The earliest surviving textiles in East Asia from Chertovy Vorota Cave, Primorye province, Russian Far East. Antiquity 86, 325–337 (2012). [Google Scholar]
11. Tanaka M., Cabrera V. M., González A. M., Larruga J. M., Takeyasu T., Fuku N., Guo L.-J., Hirose R., Fujita Y., Kurata M., Shinoda K.-i., Umetsu K., Yamada Y., Oshida Y., Sato Y., Hattori N., Mizuno Y., Arai Y., Hirose N., Ohta S., Ogawa O., Tanaka Y., Kawamori R., Shamoto-Nagai M., Maruyama W., Shimokata H., Suzuki R., Shimodaira H., Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome Res. 14, 1832–1850 (2004). [PMC free article] [PubMed] [Google Scholar]
12. Renaud G., Slon V., Duggan A. T., Kelso J., Schmutzi: Estimation of contamination and endogenous mitochondrial consensus calling for ancient DNA. Genome Biol. 16, 224 (2015). [PMC free article] [PubMed] [Google Scholar]
13. Skoglund P., Northoff B. H., Shunkov M. V., Derevianko A. P., Pääbo S., Krause J., Jakobsson M., Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc. Natl. Acad. Sci. U.S.A. 111, 2229–2234 (2014). [PMC free article] [PubMed] [Google Scholar]
14. Fu Q., Li H., Moorjani P., Jay F., Slepchenko S. M., Bondarev A. A., Johnson P. L. F., Aximu-Petri A., Prüfer K., de Filippo C., Meyer M., Zwyns N., Salazar-García D. C., Kuzmin Y. V., Keates S. G., Kosintsev P. A., Razhev D. I., Richards M. P., Peristov N. V., Lachmann M., Douka K., Higham T. F. G., Slatkin M., Hublin J.-J., Reich D., Kelso J., Bence Viola T., Pääbo S., Genome sequence of a 45,000-year-old modern human from western Siberia. Nature 514, 445–449 (2014). [PMC free article] [PubMed] [Google Scholar]
15. Raghavan M., Skoglund P., Graf K. E., Metspalu M., Albrechtsen A., Moltke I., Rasmussen S., Stafford T. W. Jr, Orlando L., Metspalu E., Karmin M., Tambets K., Rootsi S., Mägi R., Campos P. F., Balanovska E., Balanovsky O., Khusnutdinova E., Litvinov S., Osipova L. P., Fedorova S. A., Voevoda M. I., DeGiorgio M., Sicheritz-Ponten T., Brunak S., Demeshchenko S., Kivisild T., Villems R., Nielsen R., Jakobsson M., Willerslev E., Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans. Nature 505, 87–91 (2014). [PMC free article] [PubMed] [Google Scholar]
16. Allentoft M. E., Sikora M., Sjögren K.-G., Rasmussen S., Rasmussen M., Stenderup J., Damgaard P. B., Schroeder H., Ahlström T., Vinner L., Malaspinas A.-S., Margaryan A., Higham T., Chivall D., Lynnerup N., Harvig L., Baron J., Della Casa P., Dąbrowski P., Duffy P. R., Ebel A. V., Epimakhov A., Frei K., Furmanek M., Gralak T., Gromov A., Gronkiewicz S., Grupe G., Hajdu T., Jarysz R., Khartanovich V., Khokhlov A., Kiss V., Kolář J., Kriiska A., Lasak I., Longhi C., McGlynn G., Merkevicius A., Merkyte I., Metspalu M., Mkrtchyan R., Moiseyev V., Paja L., Pálfi G., Pokutta D., Pospieszny Ł., Douglas Price T., Saag L., Sablin M., Shishlina N., Smrčka V., Soenov V. I., Szeverényi V., Tóth G., Trifanova S. V., Varul L., Vicze M., Yepiskoposyan L., Zhitenev V., Orlando L., Sicheritz-Pontén T., Brunak S., Nielsen R., Kristiansen K., Willerslev E., Population genomics of Bronze Age Eurasia. Nature 522, 167–172 (2015). [PubMed] [Google Scholar]
17. Gamba C., Jones E. R., Teasdale M. D., McLaughlin R. L., Gonzalez-Fortes G., Mattiangeli V., Domboróczki L., Kővári I., Pap I., Anders A., Whittle A., Dani J., Raczky P., Higham T. F. G., Hofreiter M., Bradley D. G., Pinhasi R., Genome flux and stasis in a five millennium transect of European prehistory. Nat. Commun. 5, 5257 (2014). [PMC free article] [PubMed] [Google Scholar]
18. Patterson N., Price A. L., Reich D., Population structure and eigenanalysis. PLOS Genet. 2, e190 (2006). [PMC free article] [PubMed] [Google Scholar]
19. Alexander D. H., Novembre J., Lange K., Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 9, 1655–1664 (2009). [PMC free article] [PubMed] [Google Scholar]
20. Sánchez-Quinto F., Schroeder H., Ramirez O., Ávila-Arcos M. C., Pybus M., Olalde I., Velazquez A. M. V., Marcos M. E. P., Encinas J. M. V., Bertranpetit J., Orlando L., Gilbert M. T. P., Lalueza-Fox C., Genomic affinities of two 7,000-year-old Iberian hunter-gatherers. Curr. Biol. 22, 1494–1499 (2012). [PubMed] [Google Scholar]
21. K. Imamura, Prehistoric Japan: New Perspectives on Insular East Asia (Univ. Hawaii Press, 1996). [Google Scholar]
22. Jinam T. A., Kanzawa-Kiriyama H., Saitou N., Human genetic diversity in the Japanese Archipelago: Dual structure and beyond. Genes Genet. Syst. 90, 147–152 (2015). [PubMed] [Google Scholar]
23. Rasteiro R., Chikhi L., Revisiting the peopling of Japan: An admixture perspective. J. Hum. Genet. 54, 349–354 (2009). [PubMed] [Google Scholar]
24. He Y., Wang W. R., Xu S., Jin L.; Pan-Asia SNP Consortium, Paleolithic contingent in modern Japanese: Estimation and inference using genome-wide data. Sci. Rep. 2, 355 (2012). [PMC free article] [PubMed] [Google Scholar]
25. Jinam T. A., Kanzawa-Kiriyama H., Inoue I., Tokunaga K., Omoto K., Saitou N., Unique characteristics of the Ainu population in Northern Japan. J. Hum. Genet. 60, 565–571 (2015). [PubMed] [Google Scholar]
26. Adachi N., Shinoda K.-i., Umetsu K., Matsumura H., Mitochondrial DNA analysis of Jomon skeletons from the Funadomari site, Hokkaido, and its implication for the origins of Native American. Am. J. Phys. Anthropol. 138, 255–265 (2009). [PubMed] [Google Scholar]
27. Adachi N., Shinoda K.-i., Umetsu K., Kitano T., Matsumura H., Fujiyama R., Sawada J., Tanaka M., Mitochondrial DNA analysis of Hokkaido Jomon skeletons: Remnants of archaic maternal lineages at the southwestern edge of former Beringia. Am. J. Phys. Anthropol. 146, 346–360 (2011). [PubMed] [Google Scholar]
28. Kanzawa-Kiriyama H., Kryukov K., Jinam T. A., Hosomichi K., Saso A., Suwa G., Ueda S., Yoneda M., Tajima A., Shinoda K.-i., Inoue I., Saitou N., A partial nuclear genome of the Jomons who lived 3000 years ago in Fukushima, Japan. J. Hum. Genet. 10.1038/jhg.2016.110 (2016). [PMC free article] [PubMed] [Google Scholar]
29. HUGO Pan-Asian SNP Consortium, Mapping human genetic diversity in Asia. Science 326, 1541–1545 (2009). [PubMed] [Google Scholar]
30. Jin H.-J., Tyler-Smith C., Kim W., The peopling of Korea revealed by analyses of mitochondrial DNA and Y-chromosomal markers. PLOS ONE 4, e4210 (2009). [PMC free article] [PubMed] [Google Scholar]
31. Jin H.-J., Kwak K.-D., Hammer M. F., Nakahori Y., Shinka T., Lee J.-W., Jin F., Jia X., Tyler-Smith C., Kim W., Y-chromosomal DNA haplogroups and their implications for the dual origins of the Koreans. Hum. Genet. 114, 27–35 (2003). [PubMed] [Google Scholar]
32. Sturm R. A., Duffy D. L., Human pigmentation genes under environmental selection. Genome Biol. 13, 248 (2012). [PMC free article] [PubMed] [Google Scholar]
33. Fujimoto A., Kimura R., Ohashi J., Omi K., Yuliwulandari R., Batubara L., Mustofa M. S., Samakkarn U., Settheetham-Ishida W., Ishida T., Morishita Y., Furusawa T., Nakazawa M., Ohtsuka R., Tokunaga K., A scan for genetic determinants of human hair morphology: EDAR is associated with Asian hair thickness. Hum. Mol. Genet. 17, 835–843 (2008). [PubMed] [Google Scholar]
34. Park J.-H., Yamaguchi T., Watanabe C., Kawaguchi A., Haneji K., Takeda M., Kim Y.-I., Tomoyasu Y., Watanabe M., Oota H., Hanihara T., Ishida H., Maki K., Park S.-B., Kimura R., Effects of an Asian-specific nonsynonymous EDAR variant on multiple dental traits. J. Hum. Genet. 57, 508–514 (2012). [PubMed] [Google Scholar]
35. Itan Y., Powell A., Beaumont M. A., Burger J., Thomas M. G., The origins of lactase persistence in Europe. PLOS Comput. Biol. 5, e1000491 (2009). [PMC free article] [PubMed] [Google Scholar]
36. Crabb D. W., Edenberg H. J., Bosron W. F., Li T. K., Genotypes for aldehyde dehydrogenase deficiency and alcohol sensitivity. The inactive ALDH22 allele is dominant. J. Clin. Invest. 83, 314–316 (1989). [PMC free article] [PubMed] [Google Scholar]
37. Yang D. Y., Eng B., Waye J. S., Dudar J. C., Saunders S. R., Improved DNA extraction from ancient bones using silica-based spin columns. Am. J. Phys. Anthropol. 105, 539–543 (1998). [PubMed] [Google Scholar]
38. Meyer M., Kircher M., Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, pdb.prot5448 (2010). [PubMed] [Google Scholar]
39. Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011). [Google Scholar]
40. Renaud G., Stenzel U., Kelso J., leeHom: Adaptor trimming and merging for Illumina sequencing reads. Nucleic Acids Res. 42, e141 (2014). [PMC free article] [PubMed] [Google Scholar]
41. Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [PMC free article] [PubMed] [Google Scholar]
42. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data, The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [PMC free article] [PubMed] [Google Scholar]
43. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A., The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [PMC free article] [PubMed] [Google Scholar]
44. Jónsson H., Ginolhac A., Schubert M., Johnson P. L. F., Orlando L., mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684 (2013). [PMC free article] [PubMed] [Google Scholar]
45. Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [PMC free article] [PubMed] [Google Scholar]
46. Korneliussen T. S., Albrechtsen A., Nielsen R., ANGSD: Analysis of next generation sequencing data. BMC Bioinformatics 15, 356 (2014). [PMC free article] [PubMed] [Google Scholar]
47. Vianello D., Sevini F., Castellani G., Lomartire L., Capri M., Franceschi C., HAPLOFIND: A new method for high-throughput mtDNA haplogroup assignment. Hum. Mutat. 34, 1189–1194 (2013). [PubMed] [Google Scholar]
48. Skoglund P., Malmström H., Omrak A., Raghavan M., Valdiosera C., Günther T., Hall P., Tambets K., Parik J., Sjögren K.-G., Apel J., Willerslev E., Storå J., Götherström A., Jakobsson M., Genomic diversity and admixture differs for stone-age Scandinavian foragers and farmers. Science 344, 747–750 (2014). [PubMed] [Google Scholar]
49. Patterson N., Moorjani P., Luo Y., Mallick S., Rohland N., Zhan Y., Genschoreck T., Webster T., Reich D., Ancient admixture in human history. Genetics 192, 1065–1093 (2012). [PMC free article] [PubMed] [Google Scholar]
50. Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M. A. R., Bender D., Maller J., Sklar P., de Bakker P. I. W., Daly M. J., Sham P. C., PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [PMC free article] [PubMed] [Google Scholar]
51. Sikora M., Carpenter M. L., Moreno-Estrada A., Henn B. M., Underhill P. A., Sánchez-Quinto F., Zara I., Pitzalis M., Sidore C., Busonero F., Maschio A., Angius A., Jones C., Mendoza-Revilla J., Nekhrizov G., Dimitrova D., Theodossiev N., Harkins T. T., Keller A., Maixner F., Zink A., Abecasis G., Sanna S., Cucca F., Bustamante C. D., Population genomic analysis of ancient and modern genomes yields new insights into the genetic ancestry of the Tyrolean iceman and the genetic structure of Europe. PLOS Genet. 10, e1004353 (2014). [PMC free article] [PubMed] [Google Scholar]
52. Green R. E., Krause J., Briggs A. W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M. H.-Y., Hansen N. F., Durand E. Y., Malaspinas A.-S., Jensen J. D., Marques-Bonet T., Alkan C., Prüfer K., Meyer M., Burbano H. A., Good J. M., Schultz R., Aximu-Petri A., Butthof A., Höber B., Höffner B., Siegemund M., Weihmann A., Nusbaum C., Lander E. S., Russ C., Novod N., Affourtit J., Egholm M., Verna C., Rudan P., Brajkovic D., Kucan Ž., Gušic I., Doronichev V. B., Golovanova L. V., Lalueza-Fox C., de la Rasilla M., Fortea J., Rosas A., Schmitz R. W., Johnson P. L. F., Eichler E. E., Falush D., Birney E., Mullikin J. C., Slatkin M., Nielsen R., Kelso J., Lachmann M., Reich D., Pääbo S., A draft sequence of the Neandertal genome. Science 328, 710–722 (2010). [PMC free article] [PubMed] [Google Scholar]
53. Reich D., Thangaraj K., Patterson N., Price A. L., Singh L., Reconstructing Indian population history. Nature 461, 489–494 (2009). [PMC free article] [PubMed] [Google Scholar]
54. Browning B. L., Browning S. R., Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 98, 116–126 (2016). [PMC free article] [PubMed] [Google Scholar]
55. Andreeva Z. V., Tatarnikov V., Peshera “Chertovy Vorota v Primor’e”. Arheol. Otkrytiya 1973 Goda Cave Devils Gate Primorye 1973 Excav. Seas. Mosc., 180–181 (1974). [Google Scholar]
56. I. S. Zhushchikhovskaya, Archaeology of the Russian Far East: Essays in Stone Age Prehistory, S. M. Nelson, A. P. Derevianko, Y. V. Kuzmin, R. L. Bland, Eds. (Archaeopress, 2006), pp. 101–122. [Google Scholar]
57. Kuznecov A., The ancient settlement in a cave Devil’s Gate and some problems of the Neolithic of Primorye. Ross. Arheol. 2, 17–19 (2002). [Google Scholar]
58. Balueva T. S., Cranial samples from a Neolithic layer from Devil’s Gate Cave, Primorye. Vopr. Antropol. 58, 184–187 (1978). [Google Scholar]
59. Llorente M. G., Jones E. R., Eriksson A., Siska V., Arthur K. W., Arthur J. W., Curtis M. C., Stock J. T., Coltorti M., Pieruccini P., Stretton S., Brock F., Higham T., Park Y., Hofreiter M., Bradley D. G., Bhak J., Pinhasi R., Manica A., Ancient Ethiopian genome reveals extensive Eurasian admixture throughout the African continent. Science 350, 820–822 (2015). [PubMed] [Google Scholar]
60. Shapiro B., Hofreiter M., A paleogenomic perspective on evolution and gene function: New insights from ancient DNA. Science 343, 1236573 (2014). [PubMed] [Google Scholar]
61. Manica A., Amos W., Balloux F., Hanihara T., The effect of ancient population bottlenecks on human phenotypic variation. Nature 448, 346–348 (2007). [PMC free article] [PubMed] [Google Scholar]
62. Lamason R. L., Mohideen M.-A. P. K., Mest J. R., Wong A. C., Norton H. L., Aros M. C., Jurynec M. J., Mao X., Humphreville V. R., Humbert J. E., Sinha S., Moore J. L., Jagadeeswaran P., Zhao W., Ning G., Makalowska I., McKeigue P. M., O’Donnell D., Kittles R., Parra E. J., Mangini N. J., Grunwald D. J., Shriver M. D., Canfield V. A., Cheng K. C., SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science 310, 1782–1786 (2005). [PubMed] [Google Scholar]
63. Sturm R. A., Molecular genetics of human pigmentation diversity. Hum. Mol. Genet. 18, R9–R17 (2009). [PubMed] [Google Scholar]
64. Canfield V. A., Berg A., Peckins S., Wentzel S. M., Chung Ang K., Oppenheimer S., Cheng K. C., Molecular phylogeography of a human autosomal skin color locus under natural selection. G3 3, 2059–2067 (2013). [PMC free article] [PubMed] [Google Scholar]
65. Basu Mallick C., Mircea Iliescu F., Möls M., Hill S., Tamang R., Chaubey G., Goto R., Ho S. Y. W., Gallego Romero I., Crivellaro F., Hudjashov G., Rai N., Metspalu M., Mascie-Taylor C. G. N., Pitchappan R., Singh L., Mirazon-Lahr M., Thangaraj K., Villems R., Kivisild T., The light skin allele of SLC24A5 in south Asians and Europeans shares identity by descent. PLOS Genet. 9, e1003912 (2013). [PMC free article] [PubMed] [Google Scholar]
66. Wilde S., Timpson A., Kirsanow K., Kaiser E., Kayser M., Unterländer M., Hollfelder N., Potekhina I. D., Schier W., Thomas M. G., Burger J., Direct evidence for positive selection of skin, hair, and eye pigmentation in Europeans during the last 5,000 y. Proc. Natl. Acad. Sci. U.S.A. 111, 4832–4837 (2014). [PMC free article] [PubMed] [Google Scholar]
67. Norton H. L., Kittles R. A., Parra E., McKeigue P., Mao X., Cheng K., Canfield V. A., Bradley D. G., McEvoy B., Shriver M. D., Genetic evidence for the convergent evolution of light skin in Europeans and East Asians. Mol. Biol. Evol. 24, 710–722 (2007). [PubMed] [Google Scholar]
68. Donnelly M. P., Paschou P., Grigorenko E., Gurwitz D., Barta C., Lu R.-B., Zhukova O. V., Kim J.-J., Siniscalco M., New M., Li H., Kajuna S. L. B., Manolopoulos V. G., Speed W. C., Pakstis A. J., Kidd J. R., Kidd K. K., A global view of the OCA2-HERC2 region and pigmentation. Hum. Genet. 131, 683–696 (2011). [PMC free article] [PubMed] [Google Scholar]
69. Hider J. L., Gittelman R. M., Shah T., Edwards M., Rosenbloom A., Akey J. M., Parra E. J., Exploring signatures of positive selection in pigmentation candidate genes in populations of East Asian ancestry. BMC Evol. Biol. 13, 150 (2013). [PMC free article] [PubMed] [Google Scholar]
70. Yuasa I., Umetsu K., Harihara S., Kido A., Miyoshi A., Saitou N., Dashnyam B., Jin F., Lucotte G., Chattopadhyay P. K., Henke L., Henke J., Distribution of two Asian-related coding SNPs in the MC1R and OCA2 genes. Biochem. Genet. 45, 535–542 (2007). [PubMed] [Google Scholar]
71. Yuasa I., Umetsu K., Harihara S., Miyoshi A., Saitou N., Sook Park K., Dashnyam B., Jin F., Lucotte G., Chattopadhyay P. K., Henke L., Henke J., OCA2*481Thr, a hypofunctional allele in pigmentation, is characteristic of northeastern Asian populations. J. Hum. Genet. 52, 690–693 (2007). [PubMed] [Google Scholar]
72. Yuasa I., Harihara S., Jin F., Nishimukai H., Fujihara J., Fukumori Y., Takeshita H., Umetsu K., Saitou N., Distribution of OCA2∗481Thr and OCA2∗615Arg, associated with hypopigmentation, in several additional populations. Leg. Med. 13, 215–217 (2011). [PubMed] [Google Scholar]
73. Ramos E. M., Hoffman D., Junkins H. A., Maglott D., Phan L., Sherry S. T., Feolo M., Hindorff L. A., Phenotype–Genotype Integrator (PheGenI): Synthesizing genome-wide association study (GWAS) data with existing genomic resources. Eur. J. Hum. Genet. 22, 144–147 (2014). [PMC free article] [PubMed] [Google Scholar]
74. Fujimoto A., Ohashi J., Nishida N., Miyagawa T., Morishita Y., Tsunoda T., Kimura R., Tokunaga K., A replication study confirmed the EDAR gene to be a major contributor to population differentiation regarding head hair thickness in Asia. Hum. Genet. 124, 179–185 (2008). [PubMed] [Google Scholar]
75. Kamberov Y. G., Wang S., Tan J., Gerbault P., Wark A., Tan L., Yang Y., Li S., Tang K., Chen H., Powell A., Itan Y., Fuller D., Lohmueller J., Mao J., Schachar A., Paymer M., Hostetter E., Byrne E., Burnett M., McMahon A. P., Thomas M. G., Lieberman D. E., Jin L., Tabin C. J., Morgan B. A., Sabeti P. C., Modeling recent human evolution in mice by expression of a selected EDAR variant. Cell 152, 691–702 (2013). [PMC free article] [PubMed] [Google Scholar]
76. Goedde H. W., Agarwal D. P., Fritze G., Meier-Tackmann D., Singh S., Beckmann G., Bhatia K., Chen L. Z., Fang B., Lisker R., Paik Y. K., Rothhammer F., Saha N., Segal B., Srivastava L. M., Czeizel A., Distribution of ADH2 and ALDH2 genotypes in different populations. Hum. Genet. 88, 344–346 (1992). [PubMed] [Google Scholar]
77. Li H., Borinskaya S., Yoshimura K., Kal’ina N., Marusin A., Stepanov V. A., Qin Z., Khaliq S., Lee M.-Y., Yang Y., Mohyuddin A., Gurwitz D., Mehdi S. Q., Rogaev E., Jin L., Yankovsky N. K., Kidd J. R., Kidd K. K., Refined geographic distribution of the oriental ALDH2*504Lys (nee 487Lys) variant. Ann. Hum. Genet. 73, 335–345 (2009). [PMC free article] [PubMed] [Google Scholar]
78. Matsuo K., Hamajima N., Shinoda M., Hatooka S., Inoue M., Takezaki T., Tajima K., Gene–environment interaction between an aldehyde dehydrogenase-2 (ALDH2) polymorphism and alcohol consumption for the risk of esophageal cancer. Carcinogenesis 22, 913–916 (2001). [PubMed] [Google Scholar]
79. Cusi D., Barlassina C., Azzani T., Casari G., Citterio L., Devoto M., Glorioso N., Lanzani C., Manunta P., Righetti M., Rivera R., Stella P., Troffa C., Zagato L., Bianchi G., Polymorphisms of α-adducin and salt sensitivity in patients with essential hypertension. Lancet 349, 1353–1357 (1997). [PubMed] [Google Scholar]
80. van den Wildenberg E., Wiers R. W., Dessers J., Janssen R. G. J. H., Lambrichs E. H., Smeets H. J. M., van Breukelen G. J. P., A functional polymorphism of the μ-opioid receptor gene (OPRM1) influences cue-induced craving for alcohol in male heavy drinkers. Alcohol. Clin. Exp. Res. 31, 1–10 (2007). [PubMed] [Google Scholar]
81. Haerian B. S., Haerian M. S., OPRM1 rs1799971 polymorphism and opioid dependence: Evidence from a meta-analysis. Pharmacogenomics 14, 813–824 (2013). [PubMed] [Google Scholar]
82. Rodriguez S., Steer C. D., Farrow A., Golding J., Day I. N. M., Dependence of deodorant usage on ABCC11 genotype: Scope for personalized genetics in personal hygiene. J. Invest. Dermatol. 133, 1760–1767 (2013). [PMC free article] [PubMed] [Google Scholar]
83. Yoshiura K.-i., Kinoshita A., Ishida T., Ninokata A., Ishikawa T., Kaname T., Bannai M., Tokunaga K., Sonoda S., Komaki R., Ihara M., Saenko V. A., Alipov G. K., Sekine I., Komatsu K., Takahashi H., Nakashima M., Sosonkina N., Mapendano C. K., Ghadami M., Nomura M., Liang D.-S., Miwa N., Kim D.-K., Garidkhuu A., Natsume N., Ohta T., Tomita H., Kaneko A., Kikuchi M., Russomando G., Hirayama K., Ishibashi M., Takahashi A., Saitou N., Murray J. C., Saito S., Nakamura Y., Niikawa N., A SNP in the ABCC11 gene is the determinant of human earwax type. Nat. Genet. 38, 324–330 (2006). [PubMed] [Google Scholar]
84. Cho Y. S., Chen C.-H., Hu C., Long J., Ong R. T. H., Sim X., Takeuchi F., Wu Y., Jin Go M., Yamauchi T., Chang Y.-C., Kwak S. H., Ma R. C. W., Yamamoto K., Adair L. S., Aung T., Cai Q., Chang L.-C., Chen Y.-T., Gao Y., Hu F. B., Kim H.-L., Kim S., Kim Y. J., Lee J. J.-M., Lee N. R., Li Y., Liu J. J., Lu W., Nakamura J., Nakashima E., Ng D. P.-K., Tay W. T., Tsai F.-J., Wong T. Y., Yokota M., Zheng W., Zhang R., Wang C., So W. Y., Ohnaka K., Ikegami H., Hara K., Cho Y. M., Cho N. H., Chang T.-J., Bao Y., Hedman Å. K., Morris A. P., McCarthy M. I.; DIAGRAM Consortium; MuTHER Consortium, Takayanagi R., Park K. S., Jia W., Chuang L.-M., Chan J. C. N., Maeda S., Kadowaki T., Lee J.-Y., Wu J.-Y., Teo Y. Y., Tai E. S., Shu X. O., Mohlke K. L., Kato N., Han B.-G., Seielstad M., Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in East Asians. Nat. Genet. 44, 67–72 (2012). [PMC free article] [PubMed] [Google Scholar]