Myotis rufoniger genome sequence and analyses
Contents
[hide]- 1 Contents
- 2 붉은박쥐(Myotis rufoniger) 게놈 시퀀스 및 분석: 붉은 박쥐 게놈 특성과 윗수염박쥐 박쥐(Myotis bats)의 유효 개체수 감소
- 2.1 Associated Data
- 2.2 초록
- 2.3 소개
- 2.4 결과
- 2.5 토론
- 2.6 재료 및 방법
- 2.7 Supporting information
- 2.7.1 S1 Picture
- 2.7.2 S2 Picture
- 2.7.3 S1 Fig
- 2.7.4 S2 Fig
- 2.7.5 S3 Fig
- 2.7.6 S4 Fig
- 2.7.7 S5 Fig
- 2.7.8 S6 Fig
- 2.7.9 S1 Table
- 2.7.10 S2 Table
- 2.7.11 S3 Table
- 2.7.12 S4 Table
- 2.7.13 S5 Table
- 2.7.14 S6 Table
- 2.7.15 S7 Table
- 2.7.16 S8 Table
- 2.7.17 S9 Table
- 2.7.18 S10 Table
- 2.7.19 S11 Table
- 2.7.20 S12 Table
- 2.7.21 S13 Table
- 2.7.22 S14 Table
- 2.7.23 S15 Table
- 2.7.24 S16 Table
- 2.8 감사의 말
- 2.9 자금 명세서
- 2.10 데이터 가용성
- 2.11 참조문헌
Contents
[[[hide]]]
- 1붉은박쥐(Myotis rufoniger) 게놈 시퀀스 및 분석: 붉은 박쥐 게놈 특성과 윗수염박쥐 박쥐(Myotis bats)의 유효 개체수 감소
- 1.1Associated Data
- 1.2초록
- 1.3소개
- 1.4결과
- 1.5토론
- 1.6재료 및 방법
- 1.7Supporting information
- 1.7.1S1 Picture
- 1.7.2S2 Picture
- 1.7.3S1 Fig
- 1.7.4S2 Fig
- 1.7.5S3 Fig
- 1.7.6S4 Fig
- 1.7.7S5 Fig
- 1.7.8S6 Fig
- 1.7.9S1 Table
- 1.7.10S2 Table
- 1.7.11S3 Table
- 1.7.12S4 Table
- 1.7.13S5 Table
- 1.7.14S6 Table
- 1.7.15S7 Table
- 1.7.16S8 Table
- 1.7.17S9 Table
- 1.7.18S10 Table
- 1.7.19S11 Table
- 1.7.20S12 Table
- 1.7.21S13 Table
- 1.7.22S14 Table
- 1.7.23S15 Table
- 1.7.24S16 Table
- 1.8감사의 말
- 1.9자금 명세서
- 1.10데이터 가용성
- 1.11참조문헌
붉은박쥐(Myotis rufoniger) 게놈 시퀀스 및 분석: 붉은 박쥐 게놈 특성과 윗수염박쥐 박쥐(Myotis bats)의 유효 개체수 감소
[Author&cauthor=true&cauthor_uid=28678835 Youngjune Bhak],1,2 [Author&cauthor=true&cauthor_uid=28678835 Yeonsu Jeon],1,2 [Author&cauthor=true&cauthor_uid=28678835 Sungwon Jeon],1,2 [Author&cauthor=true&cauthor_uid=28678835 Oksung Chung],3,4 [Author&cauthor=true&cauthor_uid=28678835 Sungwoong Jho],3 [Author&cauthor=true&cauthor_uid=28678835 JeHoon Jun],3,4 [Author&cauthor=true&cauthor_uid=28678835 Hak-Min Kim],1,2 [Author&cauthor=true&cauthor_uid=28678835 Yongsoo Cho],1,2 [Author&cauthor=true&cauthor_uid=28678835 Changhan Yoon],1,5 [Author&cauthor=true&cauthor_uid=28678835 Seungwoo Lee],6 [Author&cauthor=true&cauthor_uid=28678835 Jung-Hoon Kang],7 [Author&cauthor=true&cauthor_uid=28678835 Jong-Deock Lim],7 [Author&cauthor=true&cauthor_uid=28678835 Junghwa An],8 [Author&cauthor=true&cauthor_uid=28678835 Yun Sung Cho],1,2,3,* [Author&cauthor=true&cauthor_uid=28678835 Doug-Young Ryu],6,* and [Author&cauthor=true&cauthor_uid=28678835 Jong Bhak]1,2,3,4,*
Chongle Pan, Editor
Author information Article notes Copyright and License information Disclaimer
This article has been cited by other articles in PMC.
Associated Data
초록
붉은 박쥐(Myotis rufoniger)는 윗수염박쥐속에 있는 애기박쥐과(vesper bat) 이다. 여기 우리는 붉은 박쥐(M. rufoniger) 게놈 서열과 분석을 보고한다. 우리는 66x 배의 sequencing depth에서 1.88 Gb의 추정 게놈 크기를 가진 124 Gb의 short-read DNA sequences를 생성했다. 시퀀스는 M. brandtii bat 참조 게놈에 맞춰 110× 범위 내에서 95.71%의 코딩 시퀀스 영역을 커버하는 96.50%의 매핑 속도로 정렬되었다. 윗수염박쥐속 박쥐(Myotis bat)계열의 발산시간은 1,150만 년으로 추정되며, 붉은 박쥐와 가장 가까운 종인 다비드윗수염박쥐와의 발산시간은 1,040만 년으로 추정된다. 우리는 다른 윗수염박쥐속 박쥐와 포유류 게놈에 비해 929개의 유전자에서 1,239개의 기능을 변화시키는 붉은 박쥐 특정 아미노산 서열을 발견했다. 929개의 유전자에 대한 기능적 농축 실험은 붉은 박쥐의 붉은 털 색깔과 윗수염박쥐속의 일반적인 색채에 원인이 있을 수 있는 멜라닌 관련 DCT, SLC45A2, TYRP1, OCA2 유전자의 아미노산 변화를 탐지했다. 비소 저항성(arsenic resistance)과 연관된 N6AMt1 유전자는 붉은 박쥐에서 높은 수준의 기능변화를 보였다. 우리는 또한 붉은 박쥐가 이미 박쥐의 생식, 수명, 비행, 저시력, 초음파 위치 등과 관련이 있는 것으로 이미 발표된 FSHB, GHR, IGF1R, TP53, MDM2, SLC45A2, RGS7BP, RHO, OPN1SW, CNGB3 유전자 내에 박쥐 고유 서열을 가지고 있다는 것을 확인했다. 또한 인구 통계 기록 분석을 통해 윗수염박쥐속 clade의 유효 인구 규모가 ~ 35,000 년 전부터 꾸준히 감소하고있는 것으로 나타났다. 붉은 박쥐의 유효 개체 크기는 윗수염박쥐속 박쥐 중에서 가장 낮았으며, 유전자 다양성이 상대적으로 낮다는 것을 확인하였다.
소개
붉은 박쥐(M. rufoniger)는 애기박쥐과(Vespertilionae)에 속하는 vesper bat의 한 종이다[1]. 녹슨 주황색 털(S1 Picture)로 다른 박쥐와 구별할 수 있으며, 따라서 대한민국(남한)에서는 '황금박쥐' 또는 '붉은박쥐'로 불린다. 최근에는 박쥐에 대한 분자 계통학 및 형태학 연구에 기초하여 Myotis formosus tuensis의 붉은박쥐에 학명이 재지정되었다[2]. 비록 그 개체수가 체계적으로 평가되지는 않지만, 그것은 분명히 소수의 지역에서만 수집된 희귀종이다[2]. 한국에서는 붉은박쥐가 보호되고 자연 기념물로 지정되었다. 한국에서 가장 유명하고 상징적인 보호 야생동물의 하나로, 붉은박쥐 전시관(한국 함평군 황금박쥐 전시관)도 운영되고 있다.
2012년에는 과일박쥐 Pteropus alecto와 식충성 다비드윗수염박쥐이 게놈을 발표하여 DNA 손상 체크포인트 및 DNA 복구 경로와 관련된 TP53(Tumor Protein P53)과 MDM2(MDM2 Proto-Oncogene) 유전자에 박쥐 특정 아미노산 서열을 보고하였다. 그들은 박쥐의 높은 대사율과 비행과 관련된 증가하는free radicals의 양에 대한 통찰력을 제공했다[3]. 2013년 Brandt’s bat(Myotis brandtii) 게놈 분석에서는 GHR (Growth Hormone Receptor), IGF1R (Insulin Like Growth Factor 1 Receptor), FSHB (Follicle Stimulating Hormone Beta Subunit), SLC45A2 (Solute carrier family 45, member 2), RGS7BP (Regulator of G-protein signaling 7 binding protein), RHO (Rhodopsin), OPN1SW (Opsin 1 [Cone Pigments], Short-Wave-Sensitive), CNGB3 (cyclic nucleotide gated channel beta 3) genes로, 동면 중 박쥐의 지연 배란, 긴 수명, 작은 신체 크기, 낮은 시력및 반향 위치 측정[4]에 대한 새로운 통찰력을 제공한다.
이와 같이, 가깝지만 구별되는 종의 게놈 집합은 유의한 유전자형-표현형 관련 정보를 포함할 수 있는 변이의 예측을 가능하게 한다[5–8]. 이 밀접한 종 비교 유전체학 접근법은 붉은박쥐 게놈 분석에 적용되어 붉은박쥐의 기능적, 따라서 진화적 적응을 부여할 수 있는 종 특이 변이를 식별한다. 이러한 종 특이 또는 박쥐 특이적 시퀀스 변화를 확인하고 추가로 확인하기 위해서는 서로 다른 수준의 배경 비교 종에서 가능한 한 많은 게놈으로 그러한 아미노산 변화를 평가하는 것이 중요하다. 따라서 다양한 종의 게놈의 연속적인 시퀀싱으로 생물자원을 구축하는 것이 중요하다.
붉은박쥐의 완전한 미토콘드리아 게놈은 이미 발표[9]된 반면, 아직 전체 게놈 서열은 보고되지 않아 붉은박쥐의 환경적 적응을 위한 autosomal genetic signatures 의 조사에는 한계가 있다. 더욱이, 종의 인구통계학적 역사는 깊이 배열된 전체 게놈에서만 정확하게 재구성될 수 있다[10,11]. 여기서는 유전학적 특징과 독특한 아미노산 서열을 인구학적 역사와 유전적 다양성을 동반한 대량 병렬 짧은 DNA 시퀀스(약 124Gb)를 생산하여 붉은박쥐의 전체 게놈 분석을 제공한다.
결과
붉은박쥐의 전장 유전체 분석
한국 단양 고수동굴에서 발견된 붉은박쥐 야생 사체에서 나온 게놈 DNA는 Illumina HiSeq2000 플랫폼을 이용해 시퀀싱되었다. 판독 길이 100 bp, 및 2 개의 게놈 라이브러리로부터 566 bp 및 574 bp의 표적 삽입물 크기로 총 124 Gb의 paired-end short DNA sequences 을 생성 하였다. 낮은 시퀀싱 품질 판독과 가능한 미생물 오염 판독을 줄인 후, 총 115 Gb의 DNA 시퀀스(표 1; S1 표)를 획득했다. 샘플의 종 식별을 확인하기 위해 미토콘드리아 cytochrome b sequences의 복수 시퀀스 정렬(multiple sequence alignment)을 이용하여 계통 발생 학적 분석을 실시하였으며, 우리 샘플은 붉은박쥐 (S1 Fig)에 가장 가까운 것으로 검증되었다. 붉은박쥐 전체 게놈 시퀀스를 이용하여 K-mer 분석(K = 17)을 실시하였으며, 그 게놈 크기는 대략 1.88 Gb(S2 Fig; S2 Table)로 예측되었다. 이것은 다른 박쥐 종과 비슷하고 다른 포유류의 종 보다 작다[3,4,12]. de novo assembled 붉은박쥐 게놈은 없으므로, DNA reads는 사용 가능한 세 가지 윗수염박쥐속 참조 게놈(M. brandtii, M. davidiii, M. lucifugus; 표 1; S3 표)에 모두 정렬되었다. 붉은박쥐의 일치 DNA 시퀀스는 전체 게놈 시퀀싱 데이터에서 검출된 붉은박쥐 단일염기 다형성(SNV)을 3개의 기준 게놈에 대체하여 생성되었다. 게놈 탐지율(Genome coverage, 87.63%)은 M. lucifugus 어셈블리가 참조 일 때 게놈 적용 범위 (≥10x, 87.63 %)가가장 높았고, M. davidii 게놈이 참조일때, coding sequence (CDS) coverage (≥10×, 96.60%)가 가장 높았다. SNV(58,660,193)는 M. brandtii가 참조일 때 가장 낮았고, 작은 삽입과 삭제(small insertions and deletions )는 M. daividii 가 참조일 때 가장 적었다. 그러나 M. brandtii 어셈블리가 참조일 때는 매핑율(96.50%)과 consensus genes 수(1만7247)가 가장 높았다. 따라서 M. brandtii reference-guided M. rufoniger consensus sequences를 다음의 분석에 사용했다.
Table 1
붉은 박쥐 게놈의 시퀀싱 및 매핑 통계
|
|
Myotis bat references | ||
|
M. brandtii |
M. davidii |
M. lucifugus | |
|
Sequenced reads |
1,240,507,648 | ||
|
Low quality filtered reads |
1,152,154,630 | ||
|
Contamination filtered reads |
1,149,260,980 | ||
|
Mapped reads |
1,109,045,915 |
1,101,733,443 |
1,095,435,354 |
|
Mapping rate |
96.50% |
95.86% |
95.32% |
|
Deduplicated reads |
1,023,540,144 |
1,017,982,320 |
1,010,388,456 |
|
Depth coverage |
49.5 × |
51.5 × |
49.1 × |
|
1× genome coverage |
90.17% |
87.65% |
92.29% |
|
5× genome coverage |
87.83% |
85.68% |
89.88% |
|
8× genome coverage |
86.47% |
84.60% |
88.52% |
|
10× genome coverage |
85.58% |
83.88% |
87.63% |
|
1× CDS coverage |
98.86% |
99.10% |
98.13% |
|
5× CDS coverage |
97.68% |
98.19% |
96.29% |
|
8× CDS coverage |
96.60% |
97.33% |
94.88% |
|
10× CDS coverage |
95.71% |
96.60% |
93.81% |
|
The number of homozygous SNVs |
50,615,560 |
56,635,858 |
50,441,069 |
|
The number of heterozygous SNVs |
8,044,633 |
8,008,552 |
8,306,592 |
|
The number of homozygous indels |
4,197,521 |
4,231,431 |
4,313,780 |
|
The number of heterozygous indels |
517,253 |
457,868 |
512,018 |
|
The number of consensus genes |
17,274 |
16,419 |
17,141 |
|
The number of consensus genes without gap |
12,841 |
12,618 |
12,941 |
붉은박쥐 게놈(S4 표)의 기본 구성을 조사한 결과, CDS 영역(52.15~52.95%)에서 GC 함량의 비율이 전체 게놈(41.49~42.28%)보다 높았으며, 이는 다른 포유류(다른 종의 GC 함량비 48.98~54.35%)와 일치한다. 전체 게놈에서 42.93%. S5 표). 붉은박쥐 게놈에서 알려진 transposon-derived repeats의 비율은 19.05%로 다른 박쥐(19.34~33.32%)와 견줄만 하지만 다른 포유류 게놈(25.27~51.62%, S6표)보다는 현저히 낮았다.
이 연구에 사용된 붉은박쥐 사체의 몸은 손상되었고 형태론적 성결정은 불가능했다. 따라서 X- 염색체 스캐 폴드에서의 이질성을 다른 수컷 윗수염박쥐속(S7 표)의 그것과 비교했다. 포유류 남성은 X와 Y 성 염색체를 모두 가지고 있는 반면, 여성은 2개의 X 염색체를 가지고 있기 때문에, 남성 개인은 X 염색체에서 여성 개체의 염색체보다 낮은 이질성을 보여야 한다. 우리의 샘플은 남성 다비드윗수염박쥐 개인의 그것보다 X- 염색체 스캐 폴드의 이질성이 훨씬 더 낮은 것을 보여주었는데, 이것은 분명히 남성임을 나타낸다.
붉은박쥐와 다른 포유류 종의 관계
우리는 13종의 포유류 게놈 (박쥐 게놈 7개 : Myotis brandtii, Myotis lucifugus, Myotis davidii, Eptesicus fuscus, Pteropus alecto, Pteropus vampyrus, Pteropus vampyrus, Rousettus aegyptiacus) 및 6 종의 다른 포유류 게놈 : Homo sapiens, mususuruus , Equus caballus, Heterocephalus glaber 및 Monodelphis domestica; S2 표) OrthoMCL 소프트웨어를 사용함 [13].붉은박쥐 유전자를 일치시켜 M. brandtii 유전자의 orthologous 클러스터에 추가했으며 14 종 중 6,782 개의 단일 카피 유전자 계열이 확인되었다 [7].

박쥐와 포유류 종의 계통 발생 학적 관계와 발산 시간.
추정 분기 시간 (백만 년 전; MYA)은 nodes에서 주어지며 95 % 신뢰 구간은 괄호로 묶습니다. M. brandtii—H.의 교정 시간 사피엔스 (97.5 MYA) 및 M. brandtii-P. alecto (62.6 MYA)는 TimeTree 데이터베이스에서 파생되었다.색깔의 가지와 원은 박쥐 그룹을 나타낸다 (파란색 : 곤충을 먹는 마이크로 박쥐, 빨간색 : 과일을 먹는 메가 박쥐). 외래종으로 사용 된 M. domestica는 이 그림에서 제외되었다.
붉은박쥐 특정 아미노산 시퀀스
이전 연구 [3,4]에서 FSHB, GHR, IGF1R, TP53, MDM2, SLC45A2, RGS7BP, RHO, OPN1SW 및 CNGB3 유전자 내의 박쥐 특이 적 아미노산 시퀀스가 보고되었다. 그것들은 Myotis 박쥐의 몇 가지 일반적인 특징들을 나타낸다: 지연배란(FHSB), 긴 수명(GHR과 IGF1R), 동력 비행(TP53과 MDM2), 초음파 위치(SLC45A2와 RGS7B), 그리고 저시력(RHO, OPN1SW, CNG3B). 위에 열거된 유전자 내의 동일한 아미노산 염기서열도 붉은박쥐게놈(S3 Fig)에서 확인되었다.
환경 적응 과 고유 한 진화 기능에 연계 된 박쥐 특유의 아미노산 시퀀스를 보다 더 구체적으로 식별하기 위해 붉은박쥐 특정 아미노산 변화를 다른 Myotis 박쥐와 비교 조사하였다. 붉은박쥐 에서는 2,125개의 유전자에서 총 3,366개의 독특한 아미노산 변화(uAACs)가 확인되었다. 그 중에서, 1,228개의 유전자에서 1,696개의 uAACs가 단백질 변동 효과 분석기(PROVEAN) 소프트웨어에 의해 기능이 변 할 것으로 예측되었다(변수 점수 ≤ -2.5; S8 표). [14]. 이러한 기능 변형의 변종과 관련 유전자는 붉은박쥐의 특이적 진화 적응에 대한 통찰력을 제공 할 수 있다. 갭-함유 유전자를 제외했을 때, 929 개의 유전자로부터 1,239 uAAC가 기능 변형으로 예측되었다 (S9 표). 기능 변경 uAAC를 갖는 929 개의 유전자의 기능 강화 분석은 DAVID (Annotation Visualization and Integrated Discovery) 데이터베이스 도구를 사용하여 수행되었다 [15].
아마도 기능이 변경되는 유전자는 재생 관련 용어 (EASE 점수 [Physed Fisher 's exact test]에 의한 P- 값이 0.05 이하인 Gene Ontology [GO] 분석 및 FDR (False Discovery Rate)의 10 %)가 크게 증가했습니다) 다세포 유기체에서의 생식 과정 (P- 값 : 0.00026, 39 개의 유전자, GO : 0048609), 배란주기 (P- 값 : 0.00052, 11 개의 유전자, GO : 0042698) 및 게임 생성 (P- 값 : 0.0017, 31 개의 유전자) GO : 0007276; S10 표). 붉은박쥐에서, 색소 관련 용어는 멜라닌 생합성 과정에서와 같이 기능이 변경된 유전자가 상당히enriched 해졌다 (P- 값 : 0.0037, 4 개의 유전자, GO : 0042438; S10 표).
유전자는 DCT (Dopachrome tautomerase), SLC45A2 (Solute carrier family 45), TYRP1 (Tyrosinase-related protein 1) 및 OCA2 (Oculocutaneous albinism II)이다. 우리는 다른 Myotis 박쥐의 DCT, SLC45A2, TYRP1 및 OCA2 유전자의 일부 uAAC를 PROVEAN 소프트웨어를 사용하여 기능을 변경하는 것으로 식별 할 수있었다 (표 2; S11 표) [14]. 이러한 유전자는 붉은박쥐’s 붉은 털 색깔에만 한정되지 않을음 나타낸다. 다중 시퀀스 정렬 및 특정 아미노산 시퀀스은 S4에 제시되어있다.
Table 2
멜라닌 관련 유전자 내 Myotis bat의 uAAC.
|
Gene |
Description |
The number of uAACs (The number of uAACs with PROVEAN score ≤ -2.5) | |||
|
M. rufoniger |
M. brandtii |
M. davidii |
M. lucifugus | ||
|
DCT |
Dopachrome tautomerase |
2 (2) |
0 (0) |
0 (0) |
1 (0) |
|
SLC45A2 |
Solute carrier family 45 |
3 (1) |
0 (0) |
0 (0) |
0 (0) |
|
TYRP1 |
Tyrosinase-related protein 1 |
2 (1) |
0 (0) |
1 (1) |
0 (0) |
|
OCA2 |
Oculocutaneous albinism II |
3 (3) |
1 (0) |
0 (0) |
2 (2) |
우리가 변종의 전체 유전자 기능에 관심이 있었을 때, 각 유전자의 모든 PROVEAN 변이체 점수가 합산되었다. 그런 다음 기능별 유전자 후보의 합계를 평가했다 (S12 표). 상위 20 개의 유전자에 대해, 비교를 위해 다른 Myotis 박쥐 종에서 동일한 분석이수행 되었다 (표 3; S13 및 S14 표). 우리는 변이체의 수와 합산 된 변이체 점수에 따라 붉은박쥐의 유전자 순위를 매기고 다른 Myotis 박쥐 유전자와 비교했다. 그 결과, N6AMT1 (N-6 아데닌-특이 적 DNA 메틸 트랜스퍼 라제 1)은 변이체 점수 (PROVEAN 변이체 점수의 합 : -40.285 및 변이체의 수 : 붉은박쥐에서 6 개, 다른 것에서는 uAAC가 없음) 중 네 번째로 낮은 변이체 점수를 나타냈으며, 이것은 붉은박쥐에서 N6AMT1의 높은 기능 변화를 나타낸다.
Table 3
상위 20 개 유전자에 대해 합친 PROVEAN 변이체 점수. 낮을수록 중요함.
|
Gene |
Description |
Sum of the PROVEAN variant scores | |
|
M. rufoniger |
Other Myotis bats | ||
|
BCO1 |
Beta-Carotene Oxygenase 1 |
-51.29 |
-8.13 ~ 0 |
|
CCDC184 |
Coiled-coil Domain Containing 184 |
-47.77 |
-2.88 ~ 0 |
|
PCM1 |
Pericentriolar material 1 protein |
-40.61 |
-19.04 ~ 0 |
|
N6AMT1 |
N-6 adenine-specific DNA methyltransferase 1 |
-40.29 |
0 ~ 0 |
|
MDN1 |
Midasin AAA ATPase 1 |
-37.90 |
-32.07 ~ -5.80 |
|
CEP350 |
Centrosomal protein 350 |
-37.30 |
-15.16 ~ -4.92 |
|
WNK2 |
WNK Lysine Deficient Protein Kinase 2 |
-34.67 |
-17.12 ~ -4.79 |
|
SPG11 |
Spastic Paraplegia 11 |
-29.39 |
-18.19 ~ 0 |
|
C10orf12 |
Uncharacterized protein |
-29.01 |
-18.97 ~ 0 |
|
C1orf228 |
Uncharacterized protein |
-28.71 |
-8.22 ~ 0 |
|
NOC2L |
NOC2 Like Nucleolar Associated Transcriptional Repressor |
-28.61 |
0 ~ 0 |
|
CPLX3 |
Complexin 3 |
-27.85 |
-7.67 ~ 0 |
|
KIAA1462 |
Junctional Protein Associated With Coronary Artery Disease |
-27.73 |
-3.22 ~ 0 |
|
ALPK2 |
Alpha Kinase 2 |
-26.00 |
-9.09 ~ 0 |
|
PTRH1 |
peptidyl-tRNA hydrolase 1 |
-25.30 |
0 ~ 0 |
|
MYO16 |
Myosin XVI |
-24.71 |
-14.57 ~ 0 |
|
KIAA0922 |
Transmembrane protein 131-like |
-24.59 |
-4.33 ~ 0 |
|
OAT |
Ornithine aminotransferase |
-24.44 |
-11.60 ~ 0 |
|
SPAG17 |
sperm associated antigen 17 |
-23.71 |
-16.24 ~ -2.18 |
|
IPMK |
Inositol Polyphosphate Multikinase |
-23.47 |
0 ~ 0 |
Myotis 박쥐의 개체 통계 학적 역사 및 유전 적 다양성
깊게 배열 된 게놈을 통해 집단 구조 이력의 추정 할 수 있다 [10,11]. Myotis 박쥐의 개체 통계 이력을 조사하기 위해 PSMC (Porwisewise Markovian Coalescent) 모델 추론 분석이 수행되었다 [11]. 붉은박쥐는 다른 Myotis 박쥐에 비해 약 30 ~ 300k 년 전에 가장 번성 한 종으로 추정되었으며 개체 통계 기록에 따르면 개체 수가 50 천년 전인 것으로 나타났다 (그림 2). 그러나 마지막 빙하기 (10 ~ 50k 년 전)에 개체 규모는 급격히 감소했으며 현재 가장 낮은 것으로 추정된다. 또한 ~ 30k 년 전부터 Myotis 박쥐의 유효 개체 수가 지속적으로 감소한 것으로 나타났다 (그림 2). 우리는 나아가 붉은박쥐의 게놈 다양성 (개체 규모에 영향을받을 수 있음)을 추가로 조사하여 다른 Myotis 박쥐와 비교다. heterozygous SNV 비율에 기초한 붉은박쥐 유전 적 다양성 (0.00391)은 M. davidii (0.00471) 와 M. brandtii (0.00614)보다 낮았으며, 붉은박쥐의 낮은 유효 모집단 크기를 확인했다 (S13 표 ).
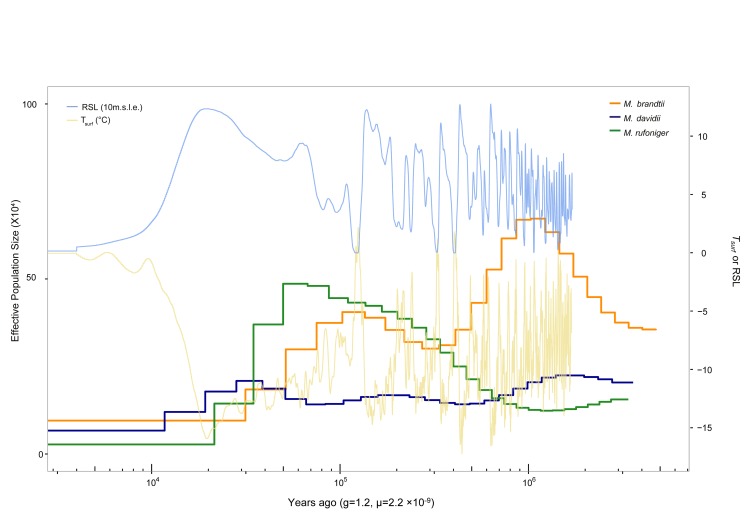
Myotis 박쥐의 개체 통계 기록.
Tsurf, 대기 표면 온도(T는 온도를 나타내고, surf는 표면을 나타냅니다), RSL, 상대 해수면, 10m.s.l., 10m 해수면 등가, g, 발생 시간(년), μ, base pair당 연간 돌연변이 비율.
토론
붉은박쥐(M. rufoniger) 유전자를 함유하는 기능 변경 변이체는 생식 관련 경로에 농축되었다. 정자 저장 연장은 Vespertilionidae (Myotis, Pipistrellus, Nyctalus, Eptesicus, Vesoertilio, Chalinolobus 및 Plecotus 속 포함) 및 Rhinolophidae과의 종 [16–18]에서 흔히 발생하는 행동이며, 따라서 복제 관련 특정 붉은박쥐가 아닐 수도 있다. 유전자는 항상 강한 자연 선택을 받고 있다. 따라서 그러한 타격은 일반적이거나 심지어 일종의 인공물로 간주될 수 있다. 따라서 재생산과 같은 기능적 범주에서 각 uAAC의 역할을 이해하려면 추가 기능 검증이 필요하다.
이전 박쥐 연구는 Myotis 박쥐의 일반적인 특징인 배란 지연 (FHSB), 긴 수명 (GHR 및 IGF1R), 동력 비행 (TP53 및 MDM2), 반향 (SLC45A2 및 RGS7B), 저시력 (RHO, OPN1SW 및 CNGB3) [3, 4]. 우리는 위에 나열된 유전자 내에서 보고된 아미노산 서열이 다른 박쥐 게놈 (S3 Fig)에서와 같이 붉은박쥐에서 보존되었다는 것을 확인했다.
우리는 또한 붉은박쥐의 유전자를 포함하는 기능 변경 독특한 변이체가 DCT, SLC45A2, TYRP1 및 OCA2 유전자를 포함한 멜라닌 관련 경로에 집중되어 있음을 발견했다. TYRP1과 DCT 유전자는 멜라닌의 양과 질에 영향을 미치는 것으로 알려져있다 [19]. TYRP1 유전자의 돌연변이는“루 포스 / 붉은 알비니즘”과 관련이있는 것으로 알려져 있으며, 적갈색 피부 나 붉은 털을 유발한다 [20-22]. OCA2 유전자에 의해 암호화된 단백질 (멜라닌 세포-특이 적 트랜스 포터 단백질)은 멜라닌 합성의 전구체 인 티로신의 트랜스포터로 보고되었다 [23]. SLC45A2 유전자 내의 변이체는 빨강 / 황색 pheomelanin 경로에 영향을 미치는 머리카락 색과의 연관성을 보여주었다 [24-25]. 이러한 맥락에서, 우리는 DCT, SLC45A2, TYRP1 및 OCA2 유전자 내의 변이체가 다른 박쥐와 구별되는 붉은박쥐의 녹슨 오렌지색 모피의 원인 일 가능성이 있다고 제안한다. 그러나 DCT, SLC45A2, TYRP1 및 OCA2 유전자 내에서 더 적지 만 특정 uAAC가 다른 Myotis 박쥐에서도 발견되어 박쥐 전체 채색에서 유전자의 일반적인 역할을 암시한다. 따라서, 착색에서 정확한 유전자-표현형 연관성을 이해하기 위해 추가 분석을 위해서는 색 다양성을 갖는 박쥐 종 세트가 필요할 것이다.
붉은박쥐 게놈에서, N6AMT1 유전자는 고도의 기능 변경을 나타냈다. N6AMT1 유전자에 의해 암호화 된 단백질 (HemK 메틸 트랜스퍼 라제 패밀리 구성원 2)은 모노 메틸 아르노 산을 디메틸 아르 신산으로 변형시켜 포유류 세포의 비소 유발 독성에 대한 내성을 부여 할 수 있다 [26]. 본 연구에서 분석된 붉은박쥐 개체의 조직에서의 원소 분석은 붉은박쥐 개체가 비소로 오염 된 음식이나 물에 노출되었음을 시사하는 장 조직에서 매우 높은 농도의 비소를 보여 주었다 [27]. 이와 관련하여, N6AMT1의 uAAC는 가능한 화학적 적응 서명으로 간주될 수 있다. 동굴은 다양한 유해 요소와 중금속과 같은 화학 물질로 인해 위험하기 때문에 동굴에 사는 박쥐에게는 일반적으로 스트레스가 많은 조건일 수 있다 [28]. 그러나 추가 실험 검증이 수행될 때까지 유해한 동굴 요소에 신속하게 적응한다는 매우 추측적인 가설이다. 또한, 이 연구는 각 종에 대해 한 사람의 게놈 데이터 만 사용했으며, 변이체 비교 분석은 개별 별 변이에 의해 편향될 수 있다. 붉은박쥐 와 Myotis 박쥐 모두에서 우리의 발견이 일반적인 적용 가능성을 확인하기 위해서는 더 많은 게놈이 시퀀싱되어야 한다.
붉은박쥐는 PSMC 분석에서 다른 Myotis 박쥐와 비교하여 가장 낮은 유효 인구 크기를 보여주었다. 이것은 그들이 상당히 널리 퍼져서 가장 위험에 처해 있음을 반드시 나타내는 것은 아니다. 가장 최근의 인간 침입에 기인하는 것이 더 합리적 일 것이다. 또한 붉은박쥐의 인구 규모는 마지막 빙하기 후반에 극적으로 감소한 것으로 나타났다. ~ 30k 년 전부터 Myotis 박쥐 가족의 유효 인구 규모가 지속적으로 감소했다. 그러나 작은 종을 사용하여 PSMC 분석을 수행했기 때문에 박쥐 전체 또는 Myotis 박쥐 특정 현상인지 확인할 수 없다. 붉은박쥐의 인구 규모의 급격한 감소는 한국 붉은박쥐의 특정 현상 일 수 있다. 따라서 박쥐의 유효 인구 규모 이력을 정확하게 모델링하려면 다양한 종 세트가 필요하다.
결론적으로, 본 연구자들은 다중 서열 정렬, uAAC 예측 변경 기능, 기능 강화 및 PSMC를 이용한 모집단 크기 추정과 같은 생물 정보학 분석을 갖는 붉은박쥐의 첫 번째 전체 게놈 서열을 보고한다. 이를 통해 박쥐가 생식 동안 장기 정자 저장, 긴 수명, 동력 비행, 저시력, 반향, 가능한 비소 저항, 모피 착색, 인구 통계 기록에서 유효 인구 규모를 지속적으로 줄인다.
재료 및 방법
샘플 및 게놈 시퀀싱
부분적으로 분해 된 붉은박쥐 시체는 2012 년 8 월 대한민국 단양의 고수 동굴(좌표 : 36 ° 59 '18.67' 'N, 128 ° 22'53.26 E; 고도 : 180.65 m)에서 고수 동굴 관리 사무실 직원에 의해 발견되었다. 붉은박쥐를 문화 재청 (National Heritage Heritage Administration)의 자연 유산 센터 (National Heritage Center)로 옮기고 얼게 유지했다 (보관 온도 : -70 ° C). 2013 년 6 월에 문화재 청의 허가하에 자연 유산 센터에서 붉은박쥐 샘플을 입수했다. 부패하지 않은 것으로 보이는 날개 막 조직은 게놈 DNA 시퀀싱을 위해 획득되었다 (S1 Picture).
게놈 DNA는 2 개의 게놈 라이브러리로부터 566 및 574 bp의 판독 길이 100 bp의 판독 길이를 갖는 Illumina HiSeq2000 플랫폼을 사용하여 시퀀싱되었다. DNA는 Tiangen Micro DNA Kit를 사용하여 추출되었다. DNA의 양은 UV 분광 광도계 (Tecan F200)를 사용하여 형광 측정법에 의해 정량화되었다. 전기 영동에 의해 부분 DNA 분해가 확인되었지만 (S2 Picture), 시퀀싱 품질 문제가 심각하지 않은 것으로 판단되었다. 게놈 DNA를 Covaris S2 Ultrasonicator를 사용하여 약 566 bp 및 574 bp로 전단 한 후 Illumina의 라이브러리 준비 프로토콜에 따라 전체 게놈 샷건 라이브러리의 준비에 사용했다. 라이브러리 구축의 각 단계의 효능은 2100 바이오 분석기를 사용하여 확인되었다 (S6 Fig). 이어서, 2 개의 라이브러리의 최종 희석을 200 사이클 동안 TruSeq PE 클러스터 키트 v3-cbot HS 및 TruSeq SBS 키트 v3-HS와 함께 HiSeq2000 시퀀서를 사용하여 시퀀싱하였다.
시퀀싱 읽기 필터링
NGSQCToolkit의 IlluQCPRLL.pl 스크립트 (ver 2.3.3; S1 Table) [29]를 사용하여 읽기 Q20 기본 함량이 70 %보다 낮을 때 DNA 읽기가 필터링되었다. 우리는 NCBI 데이터베이스에서 다른 박쥐 (Myotis brandtii, Myotis davidii, Myotis lucifugus, Pteropus Alecto 및 Pteropus vampyrus)의 게놈 데이터를 추가로 다운로드했다. 다른 박쥐로부터의 저품질 DNA 판독 또한 동일한 방법을 사용하여 여과되었다 (S1 표). 미생물 (박테리아 및 곰팡이) 게놈에 대한 판독의 정렬 점수가 Myotis 박쥐 게놈 (M. brandtii, M. davidii 및 M. lucifugus; S3 표)보다 높은 경우, 미생물 오염 된 DNA 판독이 여과되었다. 우리는 Ensembl 데이터베이스에서 다운로드 한 미생물 게놈을 사용했고 3 개의 Myotis bat 게놈 어셈블리는 NCBI에서 나왔다. DNA 판독 값은 BWA (버전 0.7.15)의 BWA-MEM 알고리즘을 사용하여 미생물 및 Myotis 박쥐 게놈에 매핑되었다. SAMtools (버전 0.1.19)의 rmdup 명령을 사용하여 PCR 복제물을 제거했다 [31]. 다른 Myotis 박쥐에서 가능한 미생물 오염 판독은 동일한 방법을 사용하여 걸러 냈지만, 자신의 게놈 어셈블리에 매핑되었다 (S1 Table).
종 식별
계통 발생 수를 구성하고 샘플의 종을 식별하기 위해, BWA의 BWA-MEM 알고리즘을 사용하여 서열 분석 데이터를 붉은박쥐 미토콘드리아 시토크롬 b 서열 (GenBank 수탁 번호 : HQ184048.1)에 맵핑함으로써 미토콘드리아 시토크롬 b 컨센서스 서열을 생성 하였다. 버전 0.7.15), 짧은 분할 조회수를 보조 옵션 (–M)으로 표시한다 [30]. 읽기 정렬 불일치를 최소화하기 위해 GATK (버전 2.5–2) RealignerTargetCreator 및 IndelRealigner 알고리즘을 사용하여 읽기를 다시 정렬했다 [32]. SAMtools (버전 0.1.19)의 rmdup 명령을 사용하여 PCR 복제물을 제거했다 [31]. 참조에 대한 전체 게놈 시퀀싱 데이터의 변형은 노이즈를 최소화하기위한 SAM 옵션 (버전 0.1.19)의 mpileup 명령을 사용하여 호출되었으며,-인서트 크기 제약과 상관없이 사용할 수 있는 옵션, -q 20 만 고려 고품질 매핑을 읽고 –Q 30은 고품질베이스만을 고려한다 [31]. 호출 된 변형은 -d 8 옵션 (최소 깊이 8)과 함께 SAMtools (ver. 0.1.19)의 vcfutils.pl varFilter 명령을 사용하여 필터링되었다 [31]. 참조에 대한 전체 게놈 서열 분석 데이터로부터 검출된 SNV는 -l 5 옵션을 갖는 SAM 도구 (ver 0.1.19)의 vcfutils.pl vcf2fq 명령을 사용하여 붉은박쥐의 미토콘드리아 시토크롬 b 컨센서스 서열을 구성하도록 대체되었다. 5 bp 창) [31]. 미토콘드리아 시토크롬 b 서열의 다중 서열 정렬은 기본 옵션 [2,33]과 함께 MUSCLE (버전 3.8.31) 프로그램을 사용하여 수행되었다. 계통 발생 학적 분석은 MEGA 7.0 프로그램을 사용하여 수행되었다 [34]. 계통 발생 수는 1,000 회 반복 부트 스트랩이있는 Tamura-Nei 모델을 기반으로 Maximum Likelihood 방법을 사용하여 추론되었다 [35,36]
게놈 크기 추정
K-mer 분석을 위해 SOAPdenovo2 패키지의 SOAPec 프로그램의 KmerFreq_HA 명령이 K = 17 옵션과 함께 사용되었다 [37]. 게놈 크기는 K-mer의 수 (깊이> 3)를 K-mer 그래프의 피크 깊이로 나눈 것으로 추정되었다.
시퀀스 데이터 매핑
필터링 된 서열화 된 판독 값은 마크가 더 짧은 스플릿 히트를 2 차 옵션으로 사용하여 BWA (버전 0.7.15)의 BWA-MEM 알고리즘을 사용하여 3 개의 Myotis bat 게놈 어셈블리 (M. brandtii, M. davidii 및 M. lucifugus)에 매핑되었다 ( -M) [30]. 읽기 정렬 불일치를 최소화하기 위해 GATK (버전 2.5–2) RealignerTargetCreator 및 IndelRealigner 알고리즘을 사용하여 읽기를 다시 정렬했다 [32]. SAMtools (버전 0.1.19)의 rmdup 명령을 사용하여 PCR 복제물을 제거했다 [31]. 붉은박쥐의 변종은 –E, -q 20, -A 및 –Q 30 옵션과 함께 SAMtools의 mpileup 명령을 사용하여 호출되었다 [31]. 호출 된 변형은 -d 8 및 -D 250 옵션 (최소 깊이 8 및 최대 깊이 250)과 함께 SAMtools의 vcfutils.pl varFilter 명령 (버전 0.1.19)을 사용하여 필터링되었다 [31]. -l 5 옵션을 사용하여 SAMtools의 vcfutils.pl vcf2fq 명령 (ver 0.1.19)을 사용하여 전체 게놈 시퀀싱 데이터에서 검출 된 붉은박쥐 SNV를 각 참조 박쥐 게놈으로 대체하여 붉은박쥐의 DNA 합의 서열을 생성했다. 31].
주석 해독
RepeatMasker (버전 4.0.5)는 기본 옵션 [38]으로 M. brandtii의 라이브러리에 대해 붉은박쥐 합의 게놈 서열을 정렬하여 이식 가능한 요소를 식별하는 데 사용되었다. 붉은박쥐의 반복 주석을 다른 종과 비교하기 위해 다른 게놈 참조에도 동일한 방법을 사용했다.
성결정
수컷 M. davidii 판독은 B 옵션 0.7-15 버전 [30]의 BWA-MEM 알고리즘을 사용하여 M. brandtii (수 샘플) 게놈 어셈블리에 매핑되었다. 읽기 불일치를 최소화하기 위해 GATK (버전 2.5–2) RealignerTargetCreator 및 IndelRealigner 알고리즘을 사용하여 읽기를 다시 정렬했다 [32]. SAMtools (버전 0.1.19)의 rmdup 명령을 사용하여 PCR 복제물을 제거했다 [31]. –E, -q 20, -A 및 –Q 30 옵션과 함께 SAMtools의 mpileup 명령을 사용하여 변형을 호출했다 [31]. 호출 된 변형은 -d 8 및 -D 250 옵션과 함께 SAMtools (ver. 0.1.19)의 vcfutils.pl varFilter 명령을 사용하여 필터링되었다 [31]. M. brandtii 게놈 어셈블리로부터 X- 염색체 스캐 폴드를 식별하기 위해, 우리는 인간의 X- 염색체 CDS 서열을 질의로 사용하여 1E-6의 E- 값 컷오프로 BLASTn (Blast 2.2.26)을 수행했다 [39]. 10 개 이상의 최고 히트를 갖는 스캐 폴드는 X- 염색체 스캐 폴드로 간주되었다.
비교 진화 분석
M. rufoniger의 서열과 13 개의 포유류 게놈 사이의 이종 상동 유전자 군 (일곱 박쥐 게놈 : M. brandtii, M. lucifugus, M. davidii, E. fuscus, P. alecto, P. vampyrus, R. aegyptiacus 및 6 개의 다른 포유류) : H. sapiens, M. musculus, B. taurus, E. caballus, H. glaber 및 M. domestica; S2 Table)은 OrthoMCL 소프트웨어 (버전 2.0.9)를 사용하여 M. rufoniger 유전자를 일치시키고 추가하여 확인했습니다. M. brandtii 유전자의 이종 클러스터 및 14 종 중 6,782 개의 단일 카피 유전자 군이 수집되었다 [7,13]. M. 브랜디, M. lucifugus, M. davidii, E. fuscus, P. alecto, P. vampyrus, R. aegyptiacus, H. sapiens, M. musculus, B. taurus, E. caballus, H. glaber 및 M 국내 게놈 및 유전자 세트는 NCBI 데이터베이스 (S3 표)로부터 다운로드되었다.계통 발생 수를 구성하고 박쥐와 다른 포유류의 발산 시간을 추정하기 위해 6,782 단일-복사 유전자 패밀리의 1,258,141 4 배 변성 부위를 MUSCLE (버전 3.8.31) 프로그램을 사용하여 다중 서열 정렬 (MSA)을 수행하는 데 사용했다. [33]. 계통 발생 수는 RAxML (Randomized Axelerated Maximum Likelihood) 프로그램 (버전 8.2)을 사용하여 14 종 중에서 구성되었다 [40]. RAxML 프로그램에서, GTR 감마 모델이 뉴클레오티드 치환 모델로 사용되었다. 분기 안정성을 확인하기 위해 1,000 개의 빠른 부트 스트랩이 사용되었다. M. domestica는 그룹 외 종으로 사용되었다. 이 계통 발생 학적 트리 토폴로지에 기초하여, 발산 시간은 MEGA 7.0 프로그램에서 Reltime-ML을 사용하여 추정되었다 [34]. 이 과정에서 M. brandtii–H 사이의 노드에서 발산 시간입니다. 사피엔스는 97.5 MYA, M. brandtii–P로 제한되었다. alecto는 TimeTree 데이터베이스를 기반으로 62.6 MYA로 제한되었다 [41].Clustal Omega (버전 1.2.4)를 사용하여 6,782 개의 단일 카피 유전자 패밀리 중에서 MSA를 구축함으로써 아미노산 변화를 확인 하였다 [42]. 기능 변경 아미노산 변화는 PROVEAN (버전 1.1.5)을 사용하여 예측되었다 [14]. DCT, SLC45A2, TYRP1, OCA2 및 N6AMT1 유전자의 MSA를 수동으로 확인하고, 잘못 정렬 된 영역 내의 uAAC를 변형 스코어 분석에서 제외시켰다. 아미노산 서열 비교 분석에서는 개별-특이 적 변이체로부터의 편향을 감소시키기 위해 동형 접합 아미노산 변이체만이 고려되었다.
인구 통계 역사 및 유전자 다양성
Myotis 박쥐의 인구 통계 기록은 PSMC (pairwise sequence Markovian coalescent) 프로그램을 사용하여 추정되었다 [11]. 우리는 Myotis bats의 다운로드 된 게놈 시퀀싱 데이터를 M 옵션을 가진 BWA 0.7.15 버전 [30]의 BWA-MEM 알고리즘을 사용하여 M. lucifugus 게놈 어셈블리에 매핑했다. 읽기 불일치를 최소화하기 위해 GATK (버전 2.5–2) RealignerTargetCreator 및 IndelRealigner 알고리즘을 사용하여 읽기를 다시 정렬했다 [32]. SAMtools (버전 0.1.19)의 rmdup 명령을 사용하여 PCR 복제물을 제거했다 [31]. SAMtools는 BAM 파일에서 이배체 게놈 정보를 추출하는 데 사용되었다 [31]. -N25 -t15 -r -p "4 + 25 * 2 + 4 + 6"의 옵션을 PSMC 분석에 사용 하였다. Myotis 박쥐의 생성 시간은 평균 성숙 및 임신 시간의 합으로 추정되었다 (S16 표) [43,44]. Myotis 박쥐의 돌연변이율은 포유류에 대해보고 된 중성 돌연변이율 (연간 염기쌍 당 2.2 × 10-9)에 생성 시간 (1.2 년)을 곱하여 추정했다 [45]. 지난 3 백만 년 동안 대기 표면 기온과 지구 상대 해수면 데이터가 얻어 져이 분석에 사용되었다 [46]. 이종 접합성 SNV의 수를 게놈 크기 (bp)로 나누어 게놈 다양성을 계산 하였다 [8]. 게놈 다양성 계산을 위해 –E, -q 20, -A 및 –Q 30 옵션과 함께 SAMtools의 mpileup 명령을 사용하여 변형을 호출했다 [31]. 호출 된 변형은 -d 8 및 -D 250 옵션과 함께 SAMtools (ver. 0.1.19)의 vcfutils.pl varFilter 명령을 사용하여 필터링되었다 [31].
Supporting information
S1 Picture
M. rufoniger carcass picture.
(JPG)
Click here for additional data file.(67K, jpg)
{kind=link}
S2 Picture
DNA sample electrophoresis.
(JPG)
Click here for additional data file.(13K, jpg)
{kind=link}
S1 Fig
Phylogenetic relationship of the M. rufoniger sample with other bats.
The phylogenetic relationship of Myotis bats was inferred from the alignment of mitochondrial cytochrome b coding sequences. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Each node has its species name and GenBank accession number.
(JPG)
Click here for additional data file.(40K, jpg)
{kind=link}
S2 Fig
Estimation of genome size using a K-mer (17-mers) analysis.
The x-axis represents depth, and the y-axis represents proportion, as calculated by the frequency at a given depth divided by the total frequency at all depths.
(PDF)
Click here for additional data file.(50K, pdf)
S3 Fig
Previously reported bats’ unique amino acid changes.
Previously reported bats’ unique amino acid sequence changes within FSHB, GHR, IGF1R, TP53, and MDM2 are highlighted in yellow; (A), Alignment of FSHB encoded peptide sequences; (B), Alignment of GHR-encoded peptide sequences; (C), Alignment of IGF1R encoded peptide sequences; (D), Alignment of TP53-encoded peptide sequences; (E), Alignment of MDM2 encoded peptide sequences; (F), Alignment of SLC45A2-encoded peptide sequences; (G), Alignment of RGS7BP-encoded peptide sequences; (H), Alignment of RHO-encoded peptide sequences; (I), Alignment of OPN1SW-encoded peptide sequences; (J), Alignment of CNGB3-encoded peptide sequences.
(PDF)
Click here for additional data file.(77K, pdf)
S4 Fig
Myotis bats’ uAACs within DCT, SLC45A2, TYRP1, and OCA2 genes.
Myotis bats’ uAACs within DCT, SLC45A2, TYRP1, and OCA2 genes are highlighted (yellow if PROVEAN score of variant ≤ -2.5, unless green). The domain region of human sequences are shaded in gray; (A) Alignment of DCT encoded peptide sequences; (B) Alignment of SLC45A2 encoded peptide sequences; (C) Alignment of TYRP1 encoded peptide sequences; (D) Alignment of OCA2 encoded peptide sequences.
(PDF)
Click here for additional data file.(467K, pdf)
S5 Fig
M. rufoniger specific amino acid sequence changes.
M. rufoniger specific amino acid sequence changes within N6AMT1 gene are highlighted in yellow.
(PDF)
Click here for additional data file.(86K, pdf)
S6 Fig
Sequencing library 574bp QC.
(A) Sequencing library 574bp QC; (B) Sequencing library 566bp QC.
(PDF)
Click here for additional data file.(154K, pdf)
S1 Table
Sequencing read filtering result.
(XLSX)
Click here for additional data file.(8.8K, xlsx)
S2 Table
17-mer statistics information.
(XLSX)
Click here for additional data file.(20K, xlsx)
S3 Table
Reference genomes used in this study.
(XLSX)
Click here for additional data file.(9.0K, xlsx)
S4 Table
Base composition of M. rufoniger genome.
The GC contents were calculated by dividing the number of [(C+G+S)–(M+K+R+Y)] ~ [(C+G+S) + (M+K+R+Y)] by the number of (sum—gap); The proportion of A, C, G, and T, and the proportion of W, S, M, K, and Y were calculated by dividing each number by the number of (sum-gap)
(XLSX)
Click here for additional data file.(12K, xlsx)
S5 Table
GC content statistics of M. rufoniger.
(XLSX)
Click here for additional data file.(11K, xlsx)
S6 Table
Transposon-derived repeats in the M. rufoniger genome.
(XLSX)
Click here for additional data file.(12K, xlsx)
S7 Table
Comparison of X-chromosomal scaffold heterozygosity.
(XLSX)
Click here for additional data file.(9.6K, xlsx)
S8 Table
M. rufoniger unique amino acid changes.
(XLSX)
Click here for additional data file.(132K, xlsx)
S9 Table
M. rufoniger unique amino acid changes from gap filtered sequences.
(XLSX)
Click here for additional data file.(120K, xlsx)
S10 Table
Functional enrichment of M. rufoniger uAACs containing genes.
(XLSX)
Click here for additional data file.(15K, xlsx)
S11 Table
PROVEAN scores of DCT, SLC45A2, TYRP1, and OCA2 genes.
(XLSX)
Click here for additional data file.(128K, xlsx)
S12 Table
Sum of the PROVEAN scores.
(XLSX)
Click here for additional data file.(127K, xlsx)
S13 Table
PROVEAN scores of top 20 ranked genes.
(XLSX)
Click here for additional data file.(140K, xlsx)
S14 Table
Sum of the PROVEAN scores of top 20 ranked genes.
(XLSX)
Click here for additional data file.(44K, xlsx)
S15 Table
Variants statistics regarding mapping of Myotis bat raw reads to the M. lucifugus reference.
(XLSX)
Click here for additional data file.(9.7K, xlsx)
S16 Table
Generation time calculation.
(XLSX)
Click here for additional data file.(11K, xlsx)
감사의 말
한국 과학 기술 정보 연구원 (KISTI)은 학술 인터넷 연결 서비스 인 한국 연구 환경 개방 네트워크 (KREONET)를 제공했다.
자금 명세서
이 작업은 울산 과학 기술원 (UNIST)의 '게놈 빅 데이터를 이용한 생물 정보학 마커 발견 분석 시스템'연구 기금 (1.150014.01)의 지원을 받았다. 또한 과학부, ICT 및 미래 계획 부 (S0503-17-1007), 게놈 연구 재단 내부 연구 기금의 PGI, 국립 생물학 연구소의 보조금 (NIBR201603103)을 통한 '소프트웨어 융합 기술 개발 프로그램'에 의해 지원되었다. 대한민국 환경부와 울산시의 게놈 한국 프로젝트가 자금을 지원하는 자료. Geromics Inc.는 OC, JHJ 및 JB에 대한 급여의 형태로 지원을 제공했지만 연구 설계, 데이터 수집 및 분석, 출판 결정 또는 원고 준비에 추가 역할을하지 않았다. 이 저자들의 구체적인 역할은 '저자 기고'섹션에 명시되어 있다.
데이터 가용성
모든 시퀀싱 파일은 NCBI (National Center for Biotechnology Information) 데이터베이스 (566bp : SRX2755014, 574bp : SRX2755088)에서 제공된다.
참조문헌